

Mitocôndria
Você sabia ...
Esta seleção wikipedia foi escolhido por voluntários que ajudam Crianças SOS da Wikipedia para este Seleção Wikipedia para as escolas. Crianças SOS tem cuidado de crianças na África por 40 anos. Você pode ajudar o seu trabalho na África ?
Em biologia celular, uma mitocôndria (mitocôndrias plural) é fechado uma membrana- organela encontrada na maioria dos eucarióticas células . Estes organelos gama de 1-10 micrómetros ( ^ M) em tamanho. As mitocôndrias são por vezes descritos como "usinas celulares" porque geram a maioria da fonte da célula de adenosina trifosfato (ATP), usado como fonte de energia química. Além de fornecer a energia celular, mitocôndrias estão envolvidas numa série de outros processos, tal como sinalização, a diferenciação celular, morte celular, bem como o controlo do ciclo celular e o crescimento celular. As mitocôndrias têm sido implicados em várias doenças humanas, incluindo transtornos mentais, disfunção cardíaca, e pode desempenhar um papel na processo de envelhecimento. A palavra vem da mitocôndria grego μίτος ou mitos, linha + χονδρίον ou khondrion, granulado. A sua descendência não é totalmente compreendida, mas, de acordo com o endosymbiotic teoria, as mitocôndrias são descendentes de antigo bactérias, que foram tragados pelos ancestrais das células eucarióticas mais de um bilhão de anos atrás.
Várias características tornam mitocôndria única. O número de mitocôndrias em uma célula varia amplamente pelo organismo e tipo de tecido. Muitas células têm apenas uma única mitocôndria, enquanto que outras podem conter vários milhares de mitocôndrias. A organela é composta de compartimentos que realizam funções especializadas. Estes compartimentos ou regiões incluem a da membrana externa, o espaço intermembranar, o membrana interna, e a cristas e matriz. Proteínas mitocondriais variam, dependendo dos tecidos e espécies. Em humanos, foram identificadas a partir mitocôndria cardíaca de 615 tipos diferentes de proteínas; Considerando que, em 940 de murino, proteínas codificadas por genes distintos foram relatados. Proteoma mitocondrial é pensado para ser regulada de forma dinâmica. Embora a maior parte do DNA de uma célula está contido no núcleo da célula, a mitocôndria tem a sua própria independente genoma. Além disso, o seu DNA mostra semelhança substancial para bacteriana genomas.
Estrutura
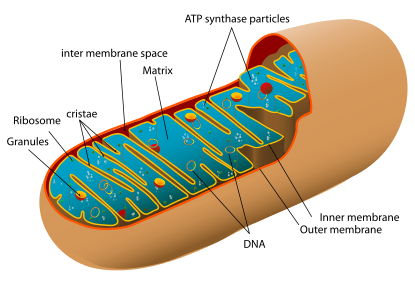
Um mitocôndria contém as membranas interiores e exteriores de compostos bicamadas de fosfolípidos e proteínas . As duas membranas, contudo, tem propriedades diferentes. Devido a esta organização de duplo membranados, existem cinco compartimentos distintos dentro da mitocôndria. Existe a membrana mitocondrial externa, o espaço intermembranar (o espaço entre as membranas exterior e interior), o membrana mitocondrial interna, o espaço de cristas (formado por invaginações da membrana interna), e a matriz (espaço dentro da membrana interna).
Membrana externa
A membrana mitocondrial externa, que inclui a totalidade organelo, tem uma proteína-a- proporção fosfolípido semelhante ao da membrana plasmática eucariótica (cerca de 1: 1 em peso). Ele contém um grande número de denominadas proteínas integrais porins. Estes porinas formar canais que permitem que moléculas de 5000 Daltons ou menos em peso molecular para livremente difundir a partir de um lado da membrana para o outro. Proteínas maiores também podem entrar na mitocôndria se uma sequência de sinalização em sua N-terminal liga-se a um grande proteína chamada múltiplas subunidades translocase da membrana externa, que, em seguida, move-os activamente através da membrana. O rompimento da membrana exterior permite que as proteínas no espaço intermembranar para vazar para o citosol, levando a morte celular determinada.
Espaço intermembranar
O espaço intermembranar é o espaço entre a membrana externa e a membrana interna. Uma vez que a membrana exterior é livremente permeável a moléculas pequenas, as concentrações de moléculas pequenas, tais como os iões e açúcares no espaço intermembranar é o mesmo que o citosol. No entanto, como grandes proteínas devem ter uma sequência de sinalização específico a ser transportados através da membrana externa, a composição proteica de este espaço é diferente do que a composição de proteína do citosol. Uma proteína que é localizada para o espaço intermembranar desta maneira é citocromo c.
Membrana interna
A membrana mitocondrial interna contém proteínas com quatro tipos de funções:
- Aqueles que executar o reacções redox de fosforilação oxidativa
- ATP sintase, que gera ATP na matriz
- Proteínas de transporte específicas que regulam metabolito passagem para dentro e para fora da matriz
- Proteína máquinas de importação.
Contém mais de 100 diferentes polipéptidos, e tem uma proporção muito elevada proteína e fosfolipido (mais do que 3: 1, em peso, que é uma proteína sobre fosfolípidos por 15). A membrana interna é o lar de cerca de 1/5 do total de proteínas em uma mitocôndria. Além disso, a membrana interna é rica em um fosfolípido incomum, cardiolipin. Este fosfolípido foi originalmente descoberto em corações de carne em 1942, e é normalmente característico das membranas plasmáticas mitocondriais e bacterianas. A cardiolipina contém quatro ácidos gordos em vez de duas e pode ajudar a tornar a membrana interior impermeável. Ao contrário da membrana externa, a membrana interior não contém porinas e é altamente impermeáveis a todas as moléculas. Quase todos os íons e moléculas requerem transportadores de membrana especiais para entrar ou sair da matriz. As proteínas são transportados para dentro da matriz através da translocase do complexo membrana interna (TIM) ou através de oxa1. Além disso, há um potencial de membrana através da membrana interna formada pela acção das enzimas da cadeia de transporte de elétrons.
Cristas
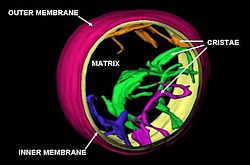
A membrana mitocondrial interna é compartimentada em numerosos cristas, que expandir a área de superfície da membrana mitocondrial interna, melhorando a sua capacidade para produzir o ATP. Estes são dobras aleatórias não simples, mas sim invaginações da membrana interna, que podem afetar global chemiosmotic função. Em típico mitocôndrias do fígado, por exemplo, a área de superfície, incluindo cristas, é cerca de cinco vezes maior do que a da membrana externa. As mitocôndrias de células que têm uma maior procura de ATP, tais como as células musculares, conter mais cristas de mitocôndrias de fígado típicos.
Matriz
A matriz é o espaço fechado pela membrana interna. Ele contém cerca de 2/3 do total de proteínas em uma mitocôndria. A matriz é importante na produção de ATP, com o auxílio da ATP sintase contido na membrana interna. A matriz contém uma mistura altamente concentrada de centenas de enzimas, mitocondrial especial ribossomas, tRNA, e várias cópias do ADN mitocondrial genoma. Das enzimas, as principais funções incluem a oxidação de piruvato e ácidos gordos , eo ciclo do ácido cítrico.
As mitocôndrias têm seu próprio material genético, e as máquinas para a fabricação de seus próprios RNAs e proteínas (ver: a biossíntese de proteínas). Uma sequência de DNA mitocondrial humano publicado revelou 16.569 pares de bases que codificam os genes totais, 37 24 tRNA e genes rRNA e 13 os genes de péptidos. A 13 mitocondrial péptidos em humanos são integrados na membrana mitocondrial interna, juntamente com as proteínas codificadas pelos genes que residem na célula hospedeira de núcleo.
Organização e distribuição
As mitocôndrias são encontrados em praticamente todos os eucariotas . Eles variam em número e localização de acordo com o tipo de célula. Um número substancial de mitocôndrias são no fígado, com cerca de 1000-2000 mitocôndrias por célula tornando-se 1/5 do volume da célula. As mitocôndrias podem ser encontrados aninhado entre myofibrils de músculo ou enrolada em torno do esperma flagelo. Muitas vezes eles formam um complexo de ramificação rede 3D dentro da célula com o citoesqueleto. A associação com o citoesqueleto determina forma mitocondrial, o que pode afectar a função bem. Evidências recentes sugerem vimentina, um dos componentes do citoesqueleto, é crítico para a associação com o citoesqueleto.
Função
Os papéis mais proeminentes da mitocôndria são sua produção de ATP e regulação de celular metabolismo. O conjunto central das reações envolvidas na produção de ATP são conhecidos coletivamente como o ciclo do ácido cítrico. No entanto, a mitocôndria tem muitas outras funções para além da produção de ATP.
Conversão de energia
Um papel dominante para a mitocôndria é a produção de ATP , tal como reflectido pelo elevado número de proteínas na membrana interna para esta tarefa. Isto é feito por oxidação dos principais produtos de glicose , piruvato, e NADH, que são produzidos no citosol. Este processo de respiração celular, também conhecido como respiração aeróbica, é dependente da presença de oxigénio . Quando o oxigénio é limitado, os produtos glicolíticas será metabolizado pela respiração anaeróbica, um processo que é independente das mitocôndrias. A produção de ATP a partir da glucose tem uma cerca de 13 vezes maior rendimento durante a respiração aeróbica em comparação com a respiração anaeróbica.
Pyruvate: o ciclo de ácido cítrico
Cada molécula de piruvato produzida pela glicólise é activamente transportada através da membrana mitocondrial interna, e para dentro da matriz, onde é oxidados e combinado com coenzima A para formar CO 2, acetil-CoA, e NADH.
A acetil-CoA é o substrato primário para introduzir o ciclo do ácido cítrico, também conhecido como o ciclo de ácido tricarboxílico (TCA) ou ciclo de Krebs. As enzimas do ciclo do ácido cítrico está localizado na matriz mitocondrial, com a excepção de succinato desidrogenase, que está ligado à membrana mitocondrial interna, como parte do complexo II. O ciclo do ácido cítrico oxida o acetil-CoA para o dióxido de carbono, e, no processo, produz cofactores reduzida (três moléculas de NADH e uma molécula de FADH 2), que são uma fonte de electrões para a cadeia de transporte de elétrons, e uma molécula de GTP (que é prontamente convertido em um ATP).
NADH e FADH 2: a cadeia de transporte de elétrons
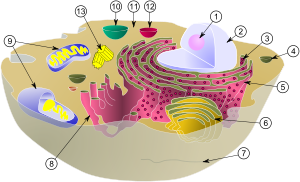
(1) nucléolo
(2) núcleo
(3) ribossomos (pequenos pontos)
(4) vesícula
(5) áspero retículo endoplasmático (ER)
(6) Aparelho de Golgi
(7) Citoesqueleto
(8) RE liso
(9) mitocôndrias
(10) vacúolo
(11) citoplasma
(12) lisossoma
(13) centrioles dentro centrossoma
A energia redox de NADH e FADH 2 é transferido para o oxigênio (O 2) em várias etapas através da cadeia de transporte de elétrons. Estas moléculas ricas em energia, são produzidos dentro da matriz através do ciclo de ácido cítrico, mas são também produzidas no citoplasma pela glicólise. Equivalentes redutores a partir do citoplasma podem ser importados por meio do malato-aspartato sistema de transporte de proteínas antiportador ou alimentar na cadeia de transporte de elétrons através de um shuttle glicerol fosfato. Complexos de proteínas na membrana interna ( NADH desidrogenase, citocromo c redutase, e citocromo-oxidase c) realizar a transferência ea liberação incremental de energia é usada para bombear protões (H +) para o espaço intermembranar. Este processo é eficiente, mas uma pequena porcentagem de elétrons pode reduzir prematuramente oxigênio, formando espécies reativas de oxigênio tais como superóxido. Isto pode causar o stress oxidativo nas mitocôndrias e podem contribuir para o declínio da função mitocondrial associada com o processo de envelhecimento.
Com o aumento da concentração de protões no espaço intermembranar, uma forte gradiente electroquímico é estabelecida através da membrana interna. Os protões pode retornar à matriz através do Complexo ATP sintase, e a sua energia potencial é usado para sintetizar ATP a partir de ADP e fosfato inorgânico (P i). Este processo é chamado quimiosmose, e foi descrito pela primeira vez por Peter Mitchell, que foi premiado com o 1978 Prêmio Nobel de Química por seu trabalho. Mais tarde, parte do Prêmio Nobel de Química 1997 foi atribuído a Paul D. Boyer e John E. Walker para seu esclarecimento do mecanismo de funcionamento do ATP sintase.
A produção de calor
Sob certas condições, protões pode voltar a entrar na matriz mitocondrial, sem contribuir para a síntese de ATP. Este processo é conhecido como vazamento de protões ou função mitocondrial e é devido à difusão facilitada de prótons na matriz. O processo resulta na energia potencial desarreados do gradiente electroquímico de protões a ser libertada na forma de calor. O processo é mediado por um canal de chamada de protões thermogenin, ou UCP1. Thermogenin é um 33k Da proteína descoberto pela primeira vez em 1973. thermogenin é encontrada principalmente em tecido adiposo marrom, ou gordura marrom, e é responsável pela termogênese sem calafrios. Tecido adiposo marrom é encontrado em mamíferos, e está em seus níveis mais elevados no início da vida e em animais que hibernam. Nos seres humanos, o tecido adiposo marrom está presente no nascimento e diminui com a idade.
Armazenamento de íons de cálcio
As concentrações de cálcio livre na célula pode regular uma variedade de reacções e é importante para transdução de sinal na célula. As mitocôndrias pode transitoriamente armazenamento de cálcio, um processo de contribuição para a homeostase da célula de cálcio. Na verdade, sua capacidade de tomar rapidamente em cálcio para a liberação mais tarde torna-os muito bons "buffers" para citossólicos cálcio. O retículo endoplasmático (ER) é o local de armazenamento mais importante de cálcio, e existe uma interacção significativa entre a mitocôndria e ER no que diz respeito ao cálcio. O cálcio é levado para cima no por uma matriz de cálcio uniporter na membrana mitocondrial interna. Ele é impulsionado principalmente pelo mitocondrial potencial de membrana. Lançamento deste cálcio de volta para o interior da célula pode ocorrer através de uma proteína troca sódio-cálcio ou através de vias "induzida por cálcio em cálcio de libertação". Isto pode iniciar picos de cálcio ou as ondas de cálcio com grandes alterações na potencial de membrana. Estes podem activar uma série de proteínas do sistema mensageiro segundo que podem coordenar processos, tais como liberação de neurotransmissores em células nervosas e liberação de hormônios em células endócrinas.
Funções adicionais
As mitocôndrias desempenham um papel central em muitos outros funções metabólicas, tais como:
- Regulamento do potencial de membrana
- Morte celular programada, a apoptose
- Excitotóxica mediada por glutamato lesão neuronal
- Regulação da proliferação celular
- Regulamento de celular metabolismo
- Certo reações de síntese do heme (Veja também: porfirina)
- Síntese de esteróides.
Algumas funções mitocondriais são realizadas somente em tipos específicos de células. Por exemplo, em mitocôndrias células do fígado contêm enzimas que permitem que eles desintoxicar amônia , um produto de resíduos do metabolismo das proteínas. Uma mutação nos genes que regulam a qualquer uma destas funções pode resultar em doenças mitocondriais.
Origem
As mitocôndrias têm muitas características em comum com procariotas. Como resultado, acredita-se ser originalmente derivados de procariontes endosymbiotic.
A mitocôndria contém DNA, que é organizado como várias cópias de um único, o cromossomo circular. Este cromossoma mitocondrial contém genes para ribossomas, e as vinte e uma ARNt do necessário para a tradução de RNAs mensageiros em proteína. A estrutura circular também é encontrada em procariotas, e a semelhança é estendido pelo facto de que o ADN mitocondrial é organizado com uma variante código genético semelhante ao do Proteobacteria. Isto sugere que o seu antepassado, a chamada proto-mitochondrion, era um membro da Proteobacteria. Em particular, o proto-mitocôndria provavelmente relacionado com a rickettsia. No entanto, a relação exacta do antepassado das mitocôndrias para o alfa-Proteobacteria e se as mitocôndrias foi formada ao mesmo tempo ou depois de o núcleo, permanece controverso.
Os ribossomas codificadas pelo ADN mitocondrial são semelhantes aos de bactérias em tamanho e estrutura. Eles se assemelham a bactérias 70S ribossoma e não o 80S ribossomas citoplasmáticos que são codificados por ADN nuclear.
O relacionamento endosymbiotic das mitocôndrias com suas células hospedeiras foi popularizada por Lynn Margulis. O endossimbiótica hipótese sugere que as mitocôndrias descendentes das bactérias que de alguma forma sobreviveram endocitose por uma outra pilha, e se tornou incorporada no citoplasma. A capacidade destas bactérias para realizar respiração em células hospedeiras que tinha invocado e glicólise fermentação teria fornecido uma vantagem evolucionária considerável. De um modo semelhante, as células hospedeiras de bactérias simbióticas capazes de fotossíntese também teria uma vantagem. A incorporação de simbiontes teria aumentado o número de ambientes em que as células poderiam sobreviver. Esta relação simbiótica provavelmente desenvolvido 1,7-2000000000 anos atrás.
Alguns grupos de eucariotas unicelulares falta mitocôndria: o microsporidians, metamonads, e archamoebae. Estes grupos aparecem como os eucariontes primitivos mais sobre árvores filogenéticas construído usando informações do rRNA, o que sugere que elas apareceram antes da origem das mitocôndrias. No entanto, isso agora é conhecido por ser um artefato de longo ramo atração - que são grupos de derivados e reter genes ou organelas derivadas da mitocôndria (por exemplo, mitosomes e hidrogenossomas).
Genoma
O genoma mitocondrial humano é uma circular de ADN de cerca de 16 molécula quilobases. Ele codifica 37 genes: para 13 subunidades de complexos respiratórios I, III, IV, e V, 22 para mitocondrial ARNt, e 2 para rRNA. Uma mitocôndria pode conter 2-10 cópias de seu DNA.
Tal como em procariontes, há uma proporção muito elevada de ADN de codificação e uma ausência de repetições. Genes mitocondriais são transcrito como multigénicos transcritos, que são clivados e poliadenilado para produzir madura mRNAs. Nem todas as proteínas necessárias para a função mitocondrial são codificados pelo genoma mitocondrial; mais são codificadas por genes no núcleo da célula e as proteínas correspondentes importados para a mitocôndria. O número exacto de genes codificados pelo núcleo ea genoma mitocondrial difere entre espécies. Em geral, os genomas mitocondriais são circulares, embora excepções foram relatados. Além disso, em geral, o ADN mitocondrial carece intrões, como é o caso no genoma mitocondrial humano; no entanto, os intrões foram observados em alguns ADN mitocondrial eucariótica, tal como a de levedura e protistas, incluindo Discoideum Dictyostelium.
Enquanto pequenas variações sobre o código padrão havia sido previsto anteriormente, nenhuma foi descoberto até 1979, quando os pesquisadores a estudar genes mitocondriais humanos determinou que eles usaram um código alternativo. Muitas variantes ligeiras têm sido descobertos desde então, incluindo vários códigos mitocondriais alternativas. Além disso, os códons AUA, AUC, AUU e são todos os codões de iniciação admissíveis.
| Organismo | Codon | Padrão | Romance |
|---|---|---|---|
| Mammalian | AGA, AGG | Arginina | Codão de terminação |
| AUA | Isoleucina | Metionina | |
| UGA | Codão de terminação | Triptofano | |
| Invertebrados | AGA, AGG | Arginina | Serina |
| AUA | Isoleucina | Metionina | |
| UGA | Codão de terminação | Triptofano | |
| Levedura | AUA | Isoleucina | Metionina |
| UGA | Codão de terminação | Triptofano | |
| CUA | Leucina | Treonina |
Algumas destas diferenças deve ser considerado como pseudo-alterações no código genético devido ao fenómeno de Edição de ARN, o que é comum nas mitocôndrias. Nas plantas superiores, pensava-se que CGG codificado para triptofano e não arginina; no entanto, o codão no RNA processado foi descoberto ser o codão UGG, consistentes com o código genético universal para o triptofano. De nota, o código genético mitocondrial artrópode sofreu evolução paralela dentro de um filo, com alguns organismos traduzindo exclusivamente AGG a lisina.
Genomas mitocondriais têm muito menos genes do que o eubact�ias a partir do qual eles são pensados para ser descendente. Embora alguns tenham sido completamente perdida, muitos foram transferidos para o núcleo, tais como os complexos de segunda respiratórias subunidades proteicas. Isto é pensado para ser relativamente comum ao longo da evolução. Alguns organismos, tais como o Cryptosporidium, realmente tem mitocôndrias que carecem de qualquer DNA, provavelmente porque todos os seus genes foram perdidos ou transferidos. Em Cryptosporidium, as mitocôndrias têm um alterada ATP sistema de geração que torna o resistente a muitos mitocondrial clássica parasita inibidores tais como o cianeto, azida, e atovaquona.
Replicação e herança
As mitocôndrias dividir por fissão binária semelhante à divisão celular bacteriana; ao contrário das bactérias, no entanto, as mitocôndrias também pode fundir-se com outras mitocôndrias .. O regulamento desta divisão difere entre eucariontes. Em muitos eucariotas unicelulares, o seu crescimento e divisão está ligada à ciclo celular. Por exemplo, um único mitocôndria podem dividir de forma síncrona com o núcleo. Este processo de divisão e de separação deve ser rigorosamente controlado de modo a que cada célula filha recebe, pelo menos, uma mitocôndria. Em outros eucariotas (por exemplo em humanos), mitocôndrias pode replicar o seu DNA e dividir essencialmente em resposta às necessidades de energia da célula, em vez de na fase com o ciclo celular. Quando as necessidades de energia de uma célula são altos, as mitocôndrias crescer e se dividir. Quando o uso de energia é baixo, as mitocôndrias são destruídas ou se tornar inativo. Em tais exemplos, e em contraste com a situação em muitos eucariotas unicelulares, as mitocôndrias são aparentemente distribuídos aleatoriamente para as células filhas durante a divisão da citoplasma.
Genes mitocondriais de um indivíduo não são herdadas pelo mesmo mecanismo como genes nucleares. Na fertilização de um óvulo por um espermatozóide, o núcleo do óvulo e esperma cada núcleo contribuem igualmente para a composição genética do núcleo zigoto. Em contraste, as mitocôndrias, e portanto, o ADN mitocondrial, vem geralmente de apenas o ovo. Mitocôndrias do esperma entrar no ovo mas não contribuir com informação genética para o embrião. Em vez disso, as mitocôndrias paternas estão marcados com ubiquitina para selecioná-los para posterior destruição no interior do embrião. A célula de ovo contém relativamente poucos mitocôndrias, mas são estas as mitocôndrias que sobrevivem e se dividem para preencher as células do organismo adulto. As mitocôndrias são, por conseguinte, na maior parte dos casos herdada para baixo da linha feminina, conhecido como herança materna. Este modo é visto na maioria dos organismos, incluindo todos os animais. No entanto, as mitocôndrias em algumas espécies podem às vezes ser herdado paternalmente. Esta é a norma entre certos plantas coníferas, embora não em pinheiros e teixos. Também tem sido sugerido que ocorre a um nível muito baixo em seres humanos.
Herança uniparental leva à pouca oportunidade para recombinação genética entre diferentes linhagens de mitocôndrias, embora uma única mitocôndria pode conter 2-10 cópias do seu ADN. Por esta razão, o ADN mitocondrial é geralmente pensado para reproduzir por fissão binária. O que recombinação tem lugar mantém a integridade genética, em vez de manter a diversidade. No entanto, há estudos que mostram evidências de recombinação do DNA mitocondrial. É claro que as enzimas necessárias para a recombinação estão presentes em células de mamíferos. Além disso, as evidências sugerem que as mitocôndrias animais podem sofrer recombinação. Os dados são um pouco mais controversa nos seres humanos, embora exista uma evidência indireta de recombinação. Se não ocorrer a recombinação, toda a sequência de ADN mitocondrial representa uma única haplótipo, o que o torna útil para o estudo da história evolutiva das populações.
Estudos genéticos de populações
A quase ausência de recombinação genética no DNA mitocondrial torna uma fonte de informação útil para os cientistas envolvidos na genética de populações e biologia evolutiva . Uma vez todo o ADN mitocondrial é herdada como uma única unidade, ou haplótipo, as relações entre o ADN mitocondrial de indivíduos diferentes podem ser representadas como um árvore gene. Patterns nestas árvores de genes pode ser usado para inferir a história evolutiva das populações. O exemplo clássico disto é em genética evolutiva humana, onde o relógio molecular pode ser usada para fornecer uma data recente para Eva mitocondrial . Isso é muitas vezes interpretado como um forte apoio para uma expansão humana moderna recente fora da África. Outro exemplo humano é o seqüenciamento do DNA mitocondrial de neandertais ossos. O relativamente grande distância evolutiva entre as sequências de DNA mitocondrial de neandertais e os seres humanos que vivem tem sido interpretado como evidência de falta de cruzamento entre os neandertais e os humanos anatomicamente-moderno.
No entanto, o DNA mitocondrial reflete a história de apenas fêmeas em uma população e por isso não pode representar a história da população como um todo. Isto pode ser parcialmente ultrapassada pela utilização de sequências genéticas paternos, como o região da recombinação não- Y-cromossomo. Num sentido mais amplo, apenas estudos que também incluem ADN nuclear pode fornecer uma história evolutiva global de uma população.
Disfunção e doença
Doenças mitocondriais
Com a sua posição central no metabolismo celular, lesão - e disfunção subsequente - em mitocôndrias é um factor importante para uma ampla gama de doenças humanas. Doenças mitocondriais frequentemente presente como distúrbios neurológicos, mas pode se manifestar como miopatia, diabetes , endocrinopatia múltipla, ou uma variedade de outras manifestações sistémicas. Doenças causadas por mutações no mtDNA incluir Síndroma de Kearns-Sayre, Síndrome de MELAS e Neuropatia óptica hereditária de Leber. Na grande maioria dos casos, estas doenças são transmitidos por uma fêmea para as suas crianças, como o zigoto deriva suas mitocôndrias e daí o seu mtDNA do óvulo. Doenças como a Síndroma de Kearns-Sayre, síndroma de Pearson, e oftalmoplegia externa progressiva são pensados para ser devido a rearranjos do mtDNA em grande escala, ao passo que outras doenças, tais como Síndrome de MELAS, Neuropatia óptica hereditária de Leber, epilepsia mioclónica com fibras vermelhas lesadas (MERRF), e outros são devido a mutações pontuais no mtDNA.
Em outras doenças, defeitos nos genes nucleares conduzem a disfunção de proteínas mitocondriais. Este é o caso em Ataxia de Friedreich, paraplegia espástica hereditária, e A doença de Wilson. Estas doenças são herdadas num relação de dominação, como se aplica à maioria das outras doenças genéticas. Uma variedade de desordens pode ser causada por mutações nucleares de enzimas de fosforilação oxidativa, tal como coenzima Q10 e deficiência Síndrome de Barth. Influências ambientais pode também interagir com predisposições hereditárias e causar doença mitocondrial. Por exemplo, pode haver uma ligação entre exposição a pesticidas eo aparecimento posterior de Doença de Parkinson.
Outras doenças não directamente ligadas às enzimas mitocondriais podem apresentar disfunção das mitocôndrias. Estes incluem esquizofrenia , transtorno bipolar, demência, doença de Alzheimer , doença de Parkinson, epilepsia , acidente vascular cerebral , doença cardiovascular, retinite pigmentosa, e diabetes mellitus . A linha comum que liga essas condições aparentemente não relacionadas é causando danos celulares estresse oxidativo e da acumulação de espécies reativas de oxigênio. Estes oxidantes então danificar o ADN mitocondrial, o que resulta na disfunção mitocondrial e morte celular.
Possíveis relações com o envelhecimento
Dado o papel das mitocôndrias como potência da célula, pode haver algum vazamento de alta energia os electrões na cadeia respiratória para formar espécies reativas de oxigênio. Isto pode resultar numa significativa estresse oxidativo nas mitocôndrias com altas taxas de mutação do DNA mitocondrial. Um ciclo vicioso é pensado para ocorrer, como o stress oxidativo leva a mutações no DNA mitocondrial, o que pode levar a modificações enzimáticas e ainda o stress oxidativo. Uma série de mudanças ocorrem a mitocôndria durante o processo de envelhecimento. Os tecidos de pacientes idosos mostram uma diminuição da actividade enzimática das proteínas de cadeia respiratória. Grandes deleções no genoma mitocondrial podem levar a elevados níveis de estresse oxidativo e morte neuronal em Doença de Parkinson. Ligações entre a hipótese de envelhecimento e estresse oxidativo não são novas e foram propostos mais de 50 anos; no entanto, há muito debate sobre se as alterações mitocondriais são causas de envelhecimento ou meramente características do envelhecimento. Um estudo notável em ratinhos não demonstrou aumento de espécies de oxigénio reactivas, apesar de aumentar mutações no DNA mitocondrial, sugerindo que o processo de envelhecimento não é devido ao stress oxidativo. Como resultado, as relações exatas entre mitocôndria, estresse oxidativo e envelhecimento ainda não foram resolvidos.