

Prion
Fundo para as escolas Wikipédia
Este conteúdo da Wikipedia foi escolhida pela SOS Children para adequação nas escolas de todo o mundo. SOS mães cada um cuidar de uma família de crianças apadrinhadas .
| Doenças de Príon (EET) | |
|---|---|
| Classificação e recursos externos | |
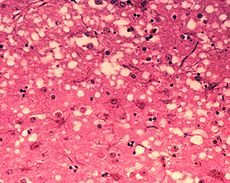 "Buracos" microscópicos são característicos em seções do tecido afetado por prião, fazendo com que o tecido para desenvolver uma arquitetura de "esponjosa". | |
| CID- 10 | A 81 |
| CID- 9 | 046 |
Um prião ( / p r Eu ɒ n /) Sob a forma do tremor epizoótico (PrPSc) é um agente infeccioso composto de proteína numa forma misfolded. Esta é a idéia central do Prion Hipótese, que continua a ser debatida. Este seria, em contraste com todos os outros agentes infecciosos conhecidos ( vírus / bactérias / fungos / parasita) que deve conter ácidos nucleicos (quer ADN , ARN, ou ambos). A palavra prion, cunhado em 1982 por Stanley B. Prusiner, é derivado da proteína de palavras e infecção. Os priões são responsáveis pela encefalopatias espongiformes transmissíveis numa variedade de mamíferos , incluindo encefalopatia espongiforme bovina (BSE, também conhecida como "doença da vaca louca") em bovinos e Doença de Creutzfeldt-Jakob (CJD) nos seres humanos. Todas as doenças de priões conhecidos afectar a estrutura do cérebro ou de outros tecido neural e todos são hoje incuráveis e universalmente fatal.
Priões propagar através da transmissão de uma estado proteína mal dobrada. Quando um príon entra em um organismo saudável, induz proteínas existentes, devidamente dobradas para converter para o, forma prião associada à doença; os atos de priões como um modelo para orientar o misfolding de mais proteínas em forma de prião. Estes prions recém-formados podem então passar a converter mais próprias proteínas; isso provoca uma reacção em cadeia que produz grandes quantidades da forma de prião. Todos os priões conhecidos induzir a formação de um dobra amilóide, em que os polimeriza proteína em um agregado que consiste de embaladas apertadamente folhas beta. Agregados amilóides são fibrilas, crescendo nas suas extremidades, e replicar quando quebra faz com que duas extremidades crescem para se tornar quatro extremidades em crescimento. O período de incubação de doenças de priões, é determinada pelo crescimento exponencial associada a taxa de replicação do prião, que é um equilíbrio entre o crescimento linear e a quebra de agregados. (Note-se que a propagação do prião depende da presença de proteína normalmente dobrada em que o prião pode induzir enrolamento incorrecto;. Os animais que não expressam a forma normal da proteína de prião não pode desenvolver nem transmitir a doença)
Esta estrutura alterada é extremamente estável e acumula-se no tecido infectado, provocando danos nos tecidos e a morte celular. Isto significa que a estabilidade estrutural priões são resistentes a desnaturação por agentes químicos e físicos, tornando eliminação e contenção destas partículas difíceis. Os priões vir em diferentes estirpes, cada uma com uma estrutura ligeiramente diferente, e a maior parte do tempo, produzir estirpes verdadeiro. Replicação Prion é, no entanto, sujeito a ocasionais epimutation e, em seguida, a seleção natural , assim como outras formas de replicação. No entanto, o número de possíveis estirpes distintas prião é provavelmente muito menor do que o número de sequências de ADN possíveis, de modo a evolução ocorre dentro de um espaço limitado.
Todas as doenças de priões, mamíferos conhecidos são causados pela chamada proteína prião, PrP. O endógeno, devidamente dobrado, forma é denotado PrP C (para C ou C COMUM ellular), enquanto a, forma misfolded ligados a doença é denotado PrP Sc (para Sc rapie, depois de uma das doenças ligadas ao primeiro priões e neurodegeneração.) A estrutura precisa do prião não é conhecido, embora possam ser formados através da combinação de PrP C, ácido poliadenilico e lípidos numa Proteína misfolding Amplificação cíclica (PMCA) reacção.
Proteínas que mostram o comportamento do tipo prião também são encontrados em alguns fungos , que tem sido útil para ajudar a compreender prions de mamíferos. Prions fúngicas não parecem causar a doença em seus hospedeiros.
Descoberta
Durante a década de 1960, dois pesquisadores com sede em Londres, biólogo radiação Tikvah Alper e matemático Stanley John Griffith surgiu a hipótese de que alguns encefalopatias espongiformes transmissíveis são causadas por um agente infeccioso que consiste exclusivamente de proteínas. Alper e Griffith desejado para explicar a descoberta de que o agente infeccioso que causa as doenças misteriosa scrapie e Doença de Creutzfeldt-Jakob resistiu radiação ionizante. (Uma única ionizante "hit" normalmente destrói toda uma partícula infecciosa, e a dose necessária para atingir metade das partículas depende do tamanho das partículas. Os dados sugerem que o agente infeccioso foi demasiado pequeno para ser um vírus.)
Francis Crick reconheceram a importância potencial da proteína-única hipótese Griffith para a propagação scrapie na segunda edição de seu " Dogma central da biologia molecular "(1970): ao afirmar que o fluxo de informações da sequência de proteína a proteína, ou a partir de proteínas de RNA e DNA foi" impedidas ", ele observou que a hipótese de Griffith era uma contradição potencial (embora não foi tão promovida por Griffith). A hipótese foi revisto mais tarde formulados, em parte, para acomodar A transcrição reversa (que tanto Howard Temin e David Baltimore descoberto em 1970).
Em 1982, Stanley B. Prusiner do University of California, San Francisco anunciou que sua equipe havia purificado o príon infeccioso hipotético, e que o agente infeccioso consistiu principalmente de uma proteína específica - embora eles não conseguiram isolar a proteína de até dois anos após o anúncio de Prusiner. Enquanto o agente infeccioso foi chamado um prião, a proteína específica que o prião foi composto por também é conhecido como o Pr ião P rotein (PrP), embora esta proteína podem ocorrer tanto nas formas infecciosas e não infecciosas. Prusiner ganhou o Prêmio Nobel de Fisiologia ou Medicina em 1997 por sua pesquisa sobre príons.
Estrutura
Isoformas
A proteína de priões que são feitas de (PRP) é encontrado por todo o corpo, mesmo em pessoas e animais saudáveis. No entanto, a PrP encontrados em material infeccioso tem uma estrutura diferente e é resistente aos proteases, as enzimas no organismo que normalmente podem quebrar as proteínas. A forma normal da proteína é denominada a PrP C, enquanto que a forma infecciosa PrPSc é chamado de - a C refere-se a "celular" ou "comum" PrP, enquanto o Sc refere-se a ' scrapie ", uma doença do prião que ocorre em ovelhas. Enquanto a PrP C é estruturalmente bem definidas, a PrPSc é certamente polydisperse e definidas a um nível relativamente pobre. PrP pode ser induzido a dobrar-se em outras isoformas mais ou menos bem definidas in vitro, e a sua relação com a forma (s) que são patogénicos in vivo ainda não é clara.
PrP C
PrP C é uma proteína normal encontrado no membranas de células . Tem 209 aminoácidos (em seres humanos), uma ligação dissulfureto, uma massa molecular de 35-36 kDa e um principalmente alfa-helicoidal estrutura. Vários existem formas topológicas; forma de superfície de uma célula ancorada via glicolípido e dois formas transmembranares. A proteína normal é não sedimentável; o que significa que não podem ser separados por técnicas de centrifugação. Sua função é uma questão complexa que continua a ser investigado. PrP C liga de cobre (II), iões com elevada afinidade. A importância deste achado não é claro, mas presumivelmente refere-se a estrutura ou função PrP. PrP C é facilmente digerida por proteinase K e pode ser libertada da superfície da célula in vitro pela enzima phosphoinositide fosfolipase C (PI-PLC), que corta o glicofosfatidilinositol (GPI) glycolipid. PrP foi relatado para desempenhar papéis importantes na adesão célula-célula e a sinalização intracelular in vivo, e podem, portanto, ser envolvida na comunicação célula a célula no cérebro.
PrPSc
O infecciosa isoforma de PrP, conhecida como PrP Sc, é capaz de converter a PrP C proteínas normais para a isoforma infecciosa, alterando a sua conformação ou forma; isto, por sua vez, altera a forma como a interconexão proteínas. Embora a estrutura exacta da PrP Sc 3D não é conhecida, tem uma maior proporção de β-estrutura de folha em vez de normal α-hélice estrutura. Agregações destas isoformas anormais forma altamente estruturada fibras amilóides, que se acumulam para formar placas. Não é claro se estes agregados são a causa de danos nas células ou são simplesmente um efeito colateral do processo de doença subjacente. A extremidade de cada fibra actua como um molde sobre o qual as moléculas de proteína podem anexar livres, permitindo que a fibra a crescer. Na maioria dos casos, apenas moléculas de PrP com uma sequência de aminoácidos idêntica à infecciosa PrPSc estão incorporadas no crescimento fibra. No entanto, a transmissão de cruzamento de espécies raras também é possível. Em um prião diferente, sup35p foi mostrado ser capaz de ser incorporado agregações existentes, mesmo quando três das cinco repetições de oligopéptidos normalmente presentes foram eliminados.
Mecanismo de replicação do prião
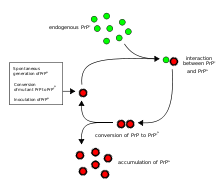
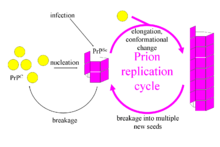
A primeira hipótese que tentou explicar como prions replicar de um modo só de proteína foi o modelo heterodímero. Este modelo assume que uma única molécula de PrP Sc se liga a uma única molécula de PrP C e catalisa a sua conversão em PrPSc. As duas moléculas PrP Sc então parta e pode ir para converter mais PrP C. No entanto, um modelo de replicação prião deve explicar tanto como priões propagar, e por sua aparência espontânea é tão raro. Manfred Eigen mostrou que o modelo heterodimer requer PrP Sc ser extraordinariamente eficaz catalisador, aumentando a taxa de conversão da reacção por um factor de cerca de 10 15. Este problema não se coloca se a PrP Sc existe apenas em formas agregadas tais como amilóide, onde cooperatividade pode agir como uma barreira para a conversão espontânea. O que é mais, apesar de um esforço considerável, infecciosa monomérica PrP Sc nunca foi isolado.
Um modelo alternativo assume que PrP Sc existe apenas como fibrilas, e que acaba de fibrilas de vincular PrP C e convertê-lo em PrP Sc. Se isso fosse tudo, então a quantidade de priões aumentaria linearmente, formando fibrilas nunca mais longos. Mas o crescimento exponencial de ambos PrPSc e do quantidade de partículas infecciosas é observada durante a doença do prião. Isto pode ser explicado tomando em consideração a quebra de fibrila. Foi encontrada uma solução matemática para a taxa de crescimento exponencial resultante da combinação de crescimento de fibrilas e fibrilas de ruptura. A taxa de crescimento exponencial depende em grande parte da raiz quadrada da concentração da PrP C. O período de incubação é determinado pela taxa de crescimento exponencial, e dados in vivo sobre as doenças de príon em camundongos transgênicos combinar esta previsão. A mesma dependência raiz quadrada também é visto em experiências in vitro com uma variedade de diferentes proteínas amilóides.
O mecanismo de replicação do prião tem implicações para a concepção de medicamentos. Uma vez que o período de incubação de doenças provocadas por priões é tão longo, uma droga eficaz não é necessário eliminar todos os priões, mas precisa apenas diminuir a taxa de crescimento exponencial. Modelos prever que a forma mais eficaz para alcançar este objectivo, usando um fármaco com a dose mais baixa possível, é o de encontrar uma droga que se liga a extremidades de fibrilas e blocos que elas cresçam ainda mais.
Função PrP
Tem sido proposto que a neurodegeneração provocadas por priões, pode ser relacionada com a função anormal da PrP. No entanto, a função fisiológica da proteína do prião continua a ser um assunto controverso. Embora os dados de experimentos in vitro sugerem muitos papéis diferentes, estudos em camundongos knockout PrP forneceram apenas informações limitadas, porque esses animais exibem apenas anormalidades menores. Na pesquisa recente feito em ratos, verificou-se que a clivagem das proteínas PrP em nervos periféricos provoca a activação de reparação de mielina em As células de Schwann e que a falta de proteínas PrP causada desmielinização nessas células.
Memória PrP e longo prazo
Uma revisão de provas, em 2005, sugeriu que PrP pode ter uma função normal na manutenção da memória de longo prazo. Como assim, um estudo de 2004 descobriu que os ratos que faltam os genes para mostrar proteína PrP celular normal alterada hipocampo potenciação a longo prazo.
PrP e renovação de células-tronco
Um artigo do Instituto Whitehead de Pesquisa Biomédica de 2006, indica que a expressão PrP com células-tronco é necessário para auto-renovação de um organismo de medula óssea. O estudo mostrou que toda a longo prazo células-tronco hematopoéticas expressa PrP em sua membrana celular e que os tecidos com células-tronco hematopoiéticas PrP nulos exibiram aumento da sensibilidade à depleção de células.
Doença de príon
| Animal afetado (s) | Doença |
|---|---|
| ovelha , cabra | Scrapie |
| gado | Encefalopatia espongiforme bovina (BSE), doença da vaca louca |
| pele de marta | Mink encefalopatia transmissível (TME) |
| veados de cauda branca, alces, veados, alces | Doença debilitante crônica (CWD) |
| gato | Encefalopatia espongiforme felina (FSE) |
| Nyala, órix, maior kudu | Encefalopatia ungulate exótica (EUE) |
| avestruz | Encefalopatia espongiforme (Não foi mostrado para ser transmissível.) |
| humano | Doença de Creutzfeldt-Jakob (CJD) |
| Doença iatrogênica de Creutzfeldt-Jakob (iCJD) | |
| Variante da doença de Creutzfeldt-Jakob (vCJD) | |
| Doença de Creutzfeldt-Jakob familial (fCJD) | |
| Doença de Creutzfeldt-Jakob esporádica (sCJD) | |
| Síndrome de Gerstmann-Sträussler-Scheinker (GSS) | |
| Insônia familiar fatal (FFI) | |
| Kuru |
Os priões causam doença neurodegenerativa por agregação extracelularmente dentro do sistema nervoso central para formar placas conhecidas como amilóide, que perturbem o normal, estrutura de tecido. Esta perturbação é caracterizada por "buracos" no tecido esponjoso com a arquitetura resultante devido à formação de vacúolo nos neurônios. Outras mudanças incluem histológicos astrogliose e a ausência de um reacção inflamatória. Enquanto o período de incubação de doenças de príon é geralmente muito longo, uma vez que os sintomas aparecem a doença progride rapidamente, levando a danos cerebrais e morte. Sintomas neurodegenerativas podem incluir convulsões, demência, ataxia (equilíbrio e disfunção coordenação), e as mudanças de comportamento ou de personalidade.
Todas as doenças de príon conhecidos, chamados coletivamente encefalopatias espongiformes transmissíveis (EET), são intratável e fatal. Uma vacina foi desenvolvido em ratinhos, no entanto, que podem fornecer informações sobre proporcionando uma vacina em humanos para resistir a infecções prião. Além disso, em 2006 os cientistas anunciaram que tinham gado geneticamente modificado falta um gene necessário para a produção de príon - assim, teoricamente, tornando-os imunes à BSE, com base em pesquisas que indicam que os ratos que faltam ocorrendo normalmente proteína príon são resistentes à infecção por proteína prion scrapie.
Muitas espécies diferentes de mamíferos podem ser afectados por doenças de priões, como a proteína prião (PrP) é muito semelhante em todos os mamíferos. Devido a pequenas diferenças de PrP entre espécies diferentes é incomum para uma doença de prion de ser transmitido de uma espécie para outra. A variante da doença do prião humano doença de Creutzfeld-Jakob, no entanto, acredita-se ser causada por um prião a qual tipicamente infecta gado, causando Encefalopatia espongiforme bovina e é transmitido através de carne contaminada.
Transmissão
Tem sido reconhecido que as doenças de priões, podem surgir de três maneiras diferentes: adquirida, familiar ou esporádica. Supõe-se frequentemente que a forma doente interage directamente com a forma normal para torná-lo reorganizar a sua estrutura. Uma ideia, a hipótese de "proteína X", que é uma proteína celular,-ainda não identificado (Proteína X) permite a conversão da PrP C e PrP Sc, trazendo uma molécula de cada um dos dois em conjunto num complexo.
A investigação actual sugere que o método primário de infecção em animais é através da ingestão. Pensa-se que os priões podem ser depositados no meio ambiente através dos restos de animais mortos e através da urina, saliva e outros fluidos corporais. Eles podem, então, permanecer no solo por ligação a argila e outros minerais.
A Universidade da Califórnia equipa de investigação, liderada pelo ganhador do Prêmio Nobel Stanley Prusiner, apresentou elementos de prova para a teoria de que a infecção pode ocorrer a partir de priões no estrume. E desde que o estrume está presente em muitas áreas circundantes reservatórios de água, bem como utilizados em muitos campos de cultivo, ele levanta a possibilidade de transmissão generalizada. Foi relatado em janeiro de 2011 que os investigadores tinham descoberto prions se espalhando através de transmissão por via aérea em partículas de aerossol, numa testes em animais experimento focando infecção scrapie em ratos de laboratório. A evidência preliminar apoiando a noção de que os priões possam ser transmitidos através da utilização de derivados de urina gonadotrofina menopáusica humana, administrado para o tratamento de infertilidade, foi publicado em 2011.
Esterilização
Partículas infecciosas que possuam ácido nucleico dependem dele para dirigir a sua replicação continuou. Os priões, no entanto, são infecciosos por seu efeito sobre versões normais da proteína. A esterilização de priões requer, portanto, a desnaturação da proteína para um estado em que a molécula não é capaz de induzir a dobragem anormal de proteínas normais. Os priões são geralmente bastante resistentes a proteases, calor , radiação, e formalina tratamentos, apesar de a sua infecciosidade pode ser reduzido por esses tratamentos. Descontaminação prião efectiva depende hidrólise de proteínas ou de redução ou destruição da estrutura terciária da proteína. Exemplos incluem branqueador, soda cáustica , e detergentes fortemente ácidas tais como LPH. 134 ° C (274 ° F) durante 18 minutos num vapor pressurizado autoclave pode não ser suficiente para desactivar o agente de doença. Esterilização de ozono está actualmente a ser estudada como um método potencial para desnaturar prion e desativação. Renaturação de um prião totalmente desnaturado de quadro infeccioso ainda não foi alcançada; no entanto, prions parcialmente desnaturado pode ser renaturada a um estado infeccioso sob certas condições artificiais.
A Organização Mundial de Saúde recomenda qualquer um dos três procedimentos a seguir para a esterilização de todos os instrumentos cirúrgicos resistentes ao calor para garantir que eles não estão contaminados com príons:
- Mergulhe em uma panela contendo 1N NaOH e calor numa autoclave de gravidade-deslocamento a 121 ° C durante 30 minutos; limpar; enxágüe em água; e em seguida, executar processos de esterilização de rotina.
- Mergulhe em 1N ou NaClO hipoclorito de sódio (20.000 partes por milhão de cloro disponível) durante 1 hora; transferir instrumentos à água; calor num autoclave de gravidade-deslocamento a 121 ° C durante 1 hora; limpar; e em seguida, executar processos de esterilização de rotina.
- Mergulhar em NaOH 1N ou hipoclorito de sódio (20,000 partes por milhão de cloro disponível) durante 1 hora; retire e lave em água, em seguida, transferir para uma panela aberta e calor em uma gravidade de deslocamento (121 ° C) ou em uma carga porosa (134 ° C) autoclave durante 1 hora; limpar; e em seguida, executar processos de esterilização de rotina.
Debate
Se priões são o agente que causa a doença ou meramente um sintoma causado por um agente diferente ainda é debatido por uma minoria de pesquisadores. As secções seguintes descrevem várias hipóteses: alguns dizem respeito à composição do agente infeccioso (proteína-somente, a proteína com outros componentes, vírus, ou outras), enquanto outros dizem respeito ao seu mecanismo de reprodução.
Proteína-única hipótese
Antes da descoberta de priões, pensou-se que todos patógenos usado ácidos nucleicos para dirigir a sua replicação. O "proteína-única hipótese" indica que uma estrutura de proteína pode replicar sem o uso de ácido nucleico. Este foi inicialmente controversa, uma vez que contradiz a dogma central da biologia molecular, que descreve o ácido nucleico tal como a forma central, das informações replicativo.
A evidência em favor da hipótese de uma única proteína inclui:
- Sem partículas de vírus, bactérias ou fungos tenham sido conclusivamente associada com doenças provocadas por priões, embora Saccharomyces cerevisiae tem sido conhecida por estar associada com priões infecciosos, mas não letais, tais como Sup35p.
- Sem ácido nucleico tem sido conclusivamente relacionado com a infecciosidade; agente é resistente à radiação ultravioleta e nucleases.
- Nenhuma resposta imunitária à infecção.
- PrPSc experimentalmente transmitido entre uma espécie e outra resulta em PrPSc com a sequência de aminoácidos da espécie de destinatários, o que sugere que a replicação do agente dador não ocorre.
- Doença do prião Familial ocorre em famílias com uma mutação no gene da PrP, e ratos com mutações PrP desenvolver doença do prião, apesar das condições controladas em que a transmissão é impedida.
- Animais falta PrP C não contrair a doença de prion.
Fatores genéticos
A gene para a proteína normal foi identificado: o Gene RPPN. Em todos os casos de doença do prião hereditárias, existe uma a mutação no gene PRNP. Muitas mutações Prnp diferentes foram identificados e estas proteínas são mais susceptíveis de dobrar em prião anormal. Embora esta descoberta coloca um buraco na hipótese do prião geral, que os priões pode apenas proteínas agregadas de aminoácidos idêntica compo. Estas mutações podem ocorrer ao longo do gene. Algumas mutações envolvem a expansão da região de repetição do octapéptido na extremidade N-terminal da PrP. Outras mutações que foram identificadas como uma causa de doença do prião herdada ocorrer nas posições 102, 117 e 198 (GSS), 178, 200, 210 e 232 (DCJ) e 178 ( Insônia Familiar Fatal, FFI). A causa da doença pode ser prião esporádica, genética, e infecciosa , ou uma combinação destes factores. Por exemplo, a fim de ter tremor epizoótico, tanto um agente infeccioso e um genótipo susceptível precisa de estar presente.
Hipótese multi-componente
Apesar de muito esforço, títulos significativos de prião infecciosidade nunca foram produzidos por redobramento moléculas PrP puros, levantando dúvidas sobre a validade da hipótese de "proteína apenas". Além disso, a hipótese de "proteína única" falha em fornecer uma explicação molecular para a capacidade de estirpes de priões, para áreas específicas do cérebro em padrões distintos. Estas deficiências, juntamente com dados experimentais adicionais, deram origem à hipótese ou "multi-componente" "variação cofator".
Em 2007, bioquímico Surachai Supattapone e seus colegas Dartmouth College produzido purificado prions infecciosos de novo a partir de componentes definidos (PrP C, lipídios co-purificado, e uma molécula sintética polianiónico). Estes investigadores também demonstraram que a molécula polianiónica necessário para a formação do prião foi incorporado selectivamente em complexos de alta afinidade com as moléculas de PrP, levando-os para a hipótese de que os priões infecciosos pode ser composto por vários componentes do hospedeiro, incluindo a PrP, de lípidos, e moléculas polianiónicos, em vez de PrPSc sozinho.
Em 2010, Jiyan Ma e seus colegas da Universidade do Estado de Ohio produziu prions infecciosos de uma receita de PrP recombinante expressa em bactérias, POPG phospholipid e RNA, apoiam a hipótese de multi-componente. Este achado está em contraste com estudos que encontraram minimamente prions infecciosos produzidos a partir sozinho recombinante PrP.
Em 2012, Supattapone e colegas purificado a fosfatidiletanolamina lipídica da membrana como um co-factor endógeno solitário capaz de facilitar a formação de elevado título priões recombinantes derivados a partir de múltiplas estirpes de priões. Relataram ainda que a co-factor essencial para a manutenção da conformação infecciosa da PrPSc e que as moléculas de cofactor ditar as propriedades de tensão de priões infecciosos.
Hipótese de envenenamento por metais pesados
Relatórios recentes sugerem que o desequilíbrio do cérebro de metal homeostase é uma causa significativa de PrP Sc -associated neurotoxicidade, embora os mecanismos subjacentes são difíceis de explicar com base na informação existente. Hipóteses propostas incluem um papel funcional para a PrP C no metabolismo de metal, e a perda de função deste devido a agregação para a doença associada a PrPSc forma como a causa do desequilíbrio cérebro de metal. Outros pontos de vista sugerem ganho de função tóxica por PrPSc devido a sequestro da PrP C -associated metais dentro dos agregados, resultando na geração de complexos de PrP Sc redox-activo. As implicações fisiológicas de algumas interações -Metal PrP C são conhecidos, enquanto outros ainda não estão claros. As implicações patológicas de PrP C interacção -Metal incluem danos oxidativos induzidos por metais, e em alguns casos a conversão da PrP C a uma -like forma PrPSc.
Hipótese viral
A hipótese só de proteína tem sido criticado por aqueles que sentem que a explicação mais simples da evidência até à data é viral. Por mais de uma década, Neuropathologist Universidade de Yale Laura Manuelidis tem vindo a propor que as doenças de príon são causados não por um não identificado vírus lento. Em janeiro de 2007, ela e seus colegas publicaram um artigo de relato de ter encontrado um vírus em 10%, ou menos, de suas células infectadas com scrapie em cultura.
A hipótese virião afirma que EET são causadas por uma molécula informacional replicável (o que é provável que seja um ácido nucleico) ligado ao PrP. Muitos EET, incluindo scrapie ea EEB, mostrar estirpes com propriedades biológicas específicas e distintas, uma característica que os apoiantes do virion sensação hipótese não é explicado por príons.
A evidência em favor de uma hipótese viral inclui:
- Variação Estirpe: diferenças na infecciosidade do prião, incubação, sintomatologia e progressão entre as espécies que se assemelha observada entre vírus, especialmente Vírus de ARN
- A longa incubação e rápido início dos sintomas se assemelha lentivírus, tais como o HIV induzida por SIDA
- As partículas virais do tipo que não aparecem para ser composta de PrP têm sido encontradas em algumas das células de linhas de células scrapie- ou infectadas com CJD.
Estudos recentes propagação infecciosidade TSE em reações livres de células e em reações químicas componente purificado sugerem fortemente contra TSE natureza viral. Mais recentemente, usando uma receita definida semelhante de componentes múltiplos (PRP, POPG lipídicas, ARN), Jiyan Ma e colegas gerado priões infecciosos de PrP recombinante expresso a partir de E. coli, lançando mais dúvidas sobre a hipótese viral.
Fungos
Proteínas fúngicas exibindo mudança conformacional modelada foram descobertos na levedura Saccharomyces cerevisiae por Reed Wickner no início de 1990. Por sua semelhança mecanicista de prions de mamíferos, eles eram chamados prions de levedura. Subsequentemente, um prião também foi encontrado no fungo Podospora anserina. Estes priões comportam-se similarmente ao PrP, mas são geralmente não tóxicos para os seus hospedeiros. O grupo de Susan Lindquist no Instituto Whitehead alegou alguns dos príons fúngicas não estão associadas a qualquer estado de doença, mas pode ter um papel útil; argumentos no entanto, os pesquisadores do NIH também ter fornecido sugerindo prions fúngicas poderia ser considerado um estado de doença. Assim, a questão de saber se as proteínas fúngicas são doenças, ou evoluíram para algumas funções específicas, ainda continua por resolver.
A partir de 2012, há oito proteínas príon conhecidos em fungos, em sete Saccharomyces cerevisiae (Sup35, Rnq1, Ure2, Swi1, Mot3, Cyc8 e Mod5) e um em Podospora anserina (HET-s). O artigo relatou que a descoberta de uma forma da proteína prião MCA1 foi recentemente retraída devido ao facto de os dados não podem ser reproduzidas. Nomeadamente, a maioria dos priões fúngicas são baseadas em sequências ricas em glutamina / asparagina, com a excepção de Het-s e Mod5.
A investigação sobre priões fúngicas deu um forte apoio ao conceito só de proteína, uma vez que a proteína purificada a partir de células extraída com um estado de prião foi demonstrada para converter a forma normal da proteína para uma forma mal dobrada in vitro, e, no processo, preservar a informação correspondente às diferentes estirpes do estado do prião. Também tem lançar alguma luz sobre domínios de priões, que são regiões em uma proteína que promovem a conversão em um prião. Prions fúngicas têm ajudado a sugerir mecanismos de conversão que podem ser aplicados a todos os prions, embora prions fúngicas aparecem distinto de prions infecciosos em mamíferos a falta de co-fator necessário para a propagação. Os domínios de priões característica pode variar entre espécies, por exemplo, domínios característicos prião fúngicas não são encontradas em mamíferos priões.
| Fúngicas Prions | |||||
|---|---|---|---|---|---|
| Proteína | Hospedeiro natural | Função normal | Estado Prion | Prion fenótipo | Ano identificado |
| Ure2p | Saccharomyces cerevisiae | Nitrogênio catabolite repressor | [URE3] | Crescimento em fontes de nitrogênio pobres | 1994 |
| Sup35p | S. cerevisiae | Factor de terminação da tradução | [PSI +] | Aumento dos níveis de supressão sem sentido | 1994 |
| HET-S | Podospora anserina | Regulamenta heterocário incompatibilidade | [Het-s] | Formação heterocário entre estirpes incompatíveis | |
| Rnq1p | S. cerevisiae | Fator modelo Protein | [Rnq +], [PIN +] | Promove agregação de outros prions | |
| MCA1 | S. cerevisiae | Putativo caspase levedura | [MCA +] | Desconhecido | 2008 |
| Swi1 | S. cerevisiae | Remodelação da cromatina | [SWI +] | O fraco crescimento em algumas fontes de carbono | 2008 |
| Cyc8 | S. cerevisiae | Repressor transcricional | [OCT +] | Desrepressão da transcrição de vários genes | 2009 |
| Mot3 | S. cerevisiae | Fator de transcrição nuclear | [MOT3 +] | Desrepressão transcricional de genes anaeróbicas | 2009 |
| SFP1 | S. cerevisiae | Fator de transcrição putativo | [ISP +] | Antisuppression | 2010 |
Potenciais tratamentos e diagnóstico
Os avanços na modelagem de computador permitiram aos cientistas para identificar compostos que podem servir como um tratamento para as doenças causadas por prião, tal como um composto que se encontra a ligar uma cavidade na PrP C e estabilizar a conformação, reduzindo a quantidade de PrPSc prejudicial.
Recentemente, anticorpos antiprion capaz de atravessar a-barreira hematoencefálica e segmentação proteína prião citosólica (uma outra forma obstáculo importante na terapêutica prião) foram descritos.
Na última década, alguns progressos foram relatados lidar com inativação de ultra-alta pressão de infecciosidade de prion em carne processada.
Em 2011, foi descoberto que os priões pode ser degradado por líquenes .
Continua a ser um problema muito prático, com diagnóstico de doenças de prion, incluindo a BSE ea DCJ. Eles têm um período de incubação de meses a décadas durante as quais não há sintomas, mesmo que o caminho de converter a proteína normal PrP cérebro para a, forma relacionada com a doença tóxica PrP Sc foi iniciado. No momento, não há praticamente nenhuma maneira de detectar PrPSc de forma confiável, exceto através da análise do cérebro usando métodos neuropatológicas e imuno-histoquímico após a morte. A acumulação de PrPSc a forma anormalmente dobrada da proteína PrP é uma característica da doença, mas está presente em níveis muito baixos em fluidos corporais facilmente acessíveis, como sangue ou urina. Os pesquisadores têm tentado desenvolver métodos para medir PrP Sc, mas ainda não há métodos totalmente aceitos para uso em materiais como o sangue.
Em 2010, uma equipe de Nova York descreveu detecção do PrP Sc mesmo quando inicialmente presente em apenas uma parte em cem mil milhões (10 -11) no tecido cerebral. O método combina a amplificação com uma nova tecnologia chamada Surround Optical Fibre Imuno (SOFIA) e alguns anticorpos específicos contra PrP Sc. Após amplificação e, em seguida, concentrando qualquer PrPSc, as amostras são marcadas com um corante fluorescente, utilizando um anticorpo para a especificidade e, em seguida, finalmente carregado para um tubo de micro-capilar. Este tubo é colocado num aparelho especialmente construído de modo que é totalmente rodeado por fibras ópticas para captar toda a luz emitida uma vez que o corante é animado utilizando um laser. A técnica permitiu a detecção da PrPSc após muitos ciclos de conversão menos do que os outros têm conseguido, reduzindo substancialmente a possibilidade de produtos manufacturados, como também acelerar o ensaio. Os pesquisadores também testaram o método em amostras de sangue de ovelhas aparentemente saudável, que passou a desenvolver scrapie. Cérebros dos animais foram analisadas uma vez quaisquer sintomas se tornou aparente. Os pesquisadores poderiam, portanto, comparar os resultados de tecido cerebral e sangue tomado uma vez os animais sintomas das doenças exibiram, com sangue obtido anteriormente na vida dos animais e de animais não infectados. Os resultados mostraram claramente que a PrPSc pode ser detectada no sangue dos animais muito antes de os sintomas aparecerem.