réaction d'aldolisation
Contexte des écoles Wikipédia
Enfants SOS offrent un chargement complet de la sélection pour les écoles pour une utilisation sur les intranets des écoles. Une bonne façon d'aider d'autres enfants est de parrainer un enfant
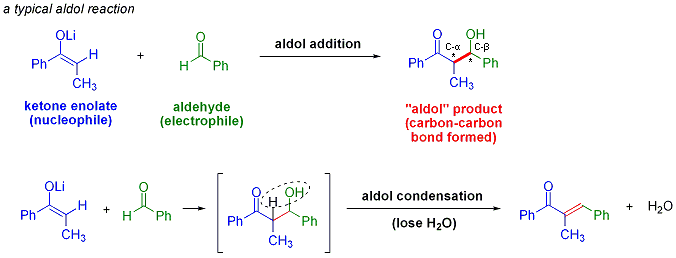
La réaction d'aldolisation est un important formation d'une liaison carbone-carbone réaction en chimie organique . Dans sa forme habituelle, il se agit de la addition nucléophile d'une cétone énolate à une aldéhyde pour former une cétone de β-hydroxy, ou "aldol" (ald ehyde + ALCOH ol) d'un motif structural trouve dans de nombreuses molécules naturelles et les produits pharmaceutiques. Parfois, le produit d'addition aldolique perd une molécule d'eau au cours de la réaction pour former un α, β-insaturée. Ceci est appelé un condensation d'aldol. La réaction d'aldol a été découvert de façon indépendante par Charles-Adolphe Wurtz et par Alexander Borodin Porfyrevich en 1872. Borodine observé la dimérisation aldolique de 3-hydroxybutanal partir l'acétaldéhyde dans des conditions acides. La réaction d'aldolisation est largement utilisé dans la production à grande échelle de produits chimiques de base tels que le pentaérythritol et le industrie pharmaceutique pour la synthèse de médicaments optiquement purs. Par exemple, route initiale de Pfizer pour le médicament Lipitor de maladie cardiaque (INN: atorvastatine), approuvé en 1996, a employé deux réactions aldoliques, permettant l'accès à des quantités multigramme échelle de la drogue.
Le motif structural aldolique est particulièrement fréquente chez polycétides, une classe de les produits naturels dont de nombreux produits pharmaceutiques sont dérivés, y compris le puissant immunosuppresseur FK506, la antibiotiques tétracyclines, et l'agent antifongique amphotéricine B. Des recherches approfondies sur la réaction aldolique a produit des méthodes hautement efficaces qui permettent au contraire difficile synthèse de nombreux polycétides en laboratoire. Ceci est important car de nombreux polycétides, avec d'autres molécules biologiquement actives, se produisent naturellement en quantités impraticable petits pour complément d'enquête. La synthèse de plusieurs de ces composés, une fois considéré presque impossible, peut maintenant être effectuée en routine à l'échelle du laboratoire, et se rapproche de la viabilité économique à plus grande échelle, dans certains cas, tels que l'agent anti-tumoral très actif discodermolide. En biochimie , la réaction d'aldolisation est une des principales étapes de glycolyse, où elle est catalysée par des enzymes appelées aldolases.
La réaction d'aldolisation est particulièrement utile dans synthèse organique car il fabrique des produits avec deux nouvelles les centres stéréogènes (sur la α- et β-carbone du produit d'addition d'aldol, marqué d'un astérisque dans le schéma ci-dessus). Les méthodes modernes, décrites ci-dessous, permettent maintenant la configuration relative et absolue de ces centres d'être contrôlé. Ceci est d'une importance particulière lors de la synthèse des produits pharmaceutiques, depuis molécules ayant la même stéréochimie connectivité de structure différente, mais ont souvent des propriétés chimiques et biologiques très différentes.
Une variété de nucléophiles peut être employé dans la réaction d'aldolisation, y compris la énols, énolates et énol éthers des cétones, des aldéhydes, et de nombreux autres composés carbonylés. Le partenaire électrophile est un aldéhyde habituellement, bien que de nombreuses variantes, comme la Réaction de Mannich, exister. Lorsque le nucléophile et électrophile sont différents (le cas habituel), la réaction est appelé une réaction aldolique croisée (par opposition à dimères formés dans une dimérisation aldolique).

Une solution de diisopropylamidure de lithium (LDA) dans le tétrahydrofuranne (THF) (dans le ballon à droite) est ajouté à une solution de propionate de tert-butyle dans le ballon à gauche, formant son énolate de lithium. Un aldéhyde peut ensuite être ajouté pour initier une réaction d'addition aldolique.
Les deux flacons sont immergés dans une glace sèche / acétone bain de refroidissement (-78 ° C) dont la température est surveillée par un thermocouple (le fil sur la gauche).
Mécanismes
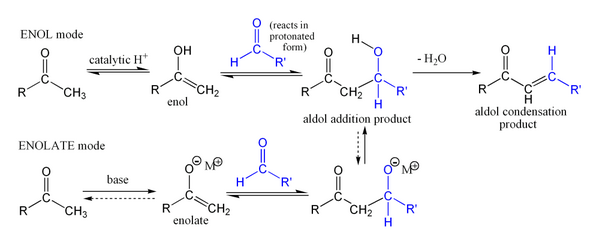
La réaction d'aldolisation peut procéder par deux mécanismes fondamentalement différents. Les composés carbonylés tels que des aldéhydes et des cétones, peuvent être convertis en énols ou des éthers d'énols. Ces composés, étant nucléophile au α-carbone, peut attaquer carbonyles protonés particulièrement réactifs tels que les aldéhydes protonés. Ce est le "mécanisme d'énol". Les composés carbonylés, étant des acides de carbone, peuvent également être déprotoné pour former des énolates, qui sont beaucoup plus fortement nucléophiles que les énols ou des éthers d'énols et des électrophiles peuvent attaquer directement. L'électrophile est un aldéhyde habituel, depuis les cétones sont beaucoup moins réactif. Ce est le «mécanisme énolate".
Si les conditions sont particulièrement dures (par exemple, NaOMe, MeOH, reflux), de la condensation peut se produire, mais cela peut généralement être évités avec des réactifs doux et des températures basses (par exemple, LDA (une base forte), le THF, -78 ° C). Bien que l'addition aldolique procède généralement à proximité de réalisation, la réaction ne est pas irréversible, étant donné que le traitement de produits d'addition d'aldol avec les bases fortes induit habituellement rétro-aldol clivage (donne les produits de départ). condensations aldoliques sont irréversibles.
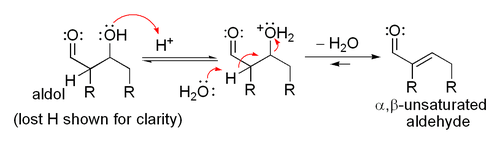
Mécanisme de Enol
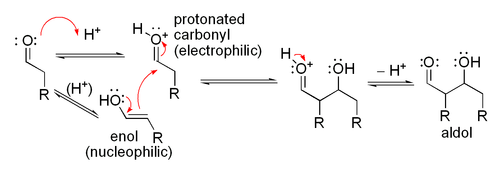
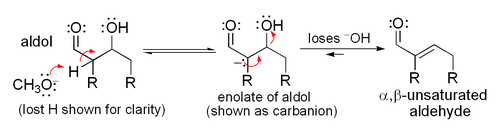
Lorsqu'un catalyseur acide est utilisé, l'étape initiale dans la Mécanisme de réaction catalysée par un acide comprend tautomérisation du composé carbonyle à l'énol. L'acide sert aussi à activer le groupe carbonyle d'une autre molécule par protonation, rendant très électrophile. L'énol est nucléophile à l'α-carbone, ce qui lui permet d'attaquer le composé carbonyle protonée, conduisant à l'aldol après déprotonation. Ce général déshydrate pour donner le composé carbonyle insaturé. Le schéma montre une auto-condensation catalysée par un acide d'un aldéhyde typique.
Aldol catalysée par un acide mécanisme
La déshydratation catalysée par un acide
Mécanisme énolate
Si le catalyseur est une base modérée telle que l'hydroxyde ou un ion un alcoxyde, la réaction d'aldolisation se fait par attaque nucléophile par la stabilisé par résonance énolate sur le groupe carbonyle d'une autre molécule. Le produit est le sel alcoolate du produit d'aldolisation. L'aldol se est alors formé, et il peut ensuite subir une déshydratation pour donner le composé carbonyle insaturé. Le schéma montre un mécanisme simple pour la base catalysée aldol réaction d'un aldéhyde avec lui-même.
Base de réaction catalysée aldolique (montré à l'aide - OCH 3 comme base)
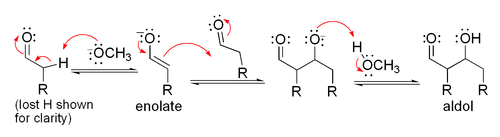
Base de déshydratation catalysée (parfois écrit comme une seule étape)
Bien que seule une quantité catalytique de base est nécessaire, dans certains cas, la procédure la plus courante consiste à utiliser un quantité stoechiométrique d'une base forte telle que LDA ou NaHMDS. Dans ce cas, la formation de l'énolate est irréversible, et le produit d'aldolisation est pas formée jusqu'à ce que l'alcoxyde de métal du produit d'aldolisation est protoné dans une étape de traitement final séparé.
Zimmerman-Traxler modèle
Des formes plus raffinées du mécanisme sont connus. En 1957, Zimmerman et Traxler proposé que certaines réactions aldoliques ont «état de transition de six chaînons s ayant une conformation chaise. "Ce est maintenant connu comme le modèle Zimmerman-Traxler. E-énolates donnent lieu à produits anti, tandis que Z-énolates donnent lieu à produits syn. Les facteurs qui contrôlent la sélectivité sont la préférence pour placer substituants équatoriale dans six chaînons états de transition et l'évitement de interactions syn-pentane, respectivement. E et Z se réfèrent à la relation stéréochimique cis-trans entre l'oxygène énolate portant le contre-ion positif et le groupe de priorité la plus élevée sur l'atome de carbone alpha. En réalité, seuls certains métaux tels que le lithium et le bore suivre de manière fiable le modèle Zimmerman-Traxler. Ainsi, dans certains cas, la résultat stéréochimique de la réaction peut être imprévisible.
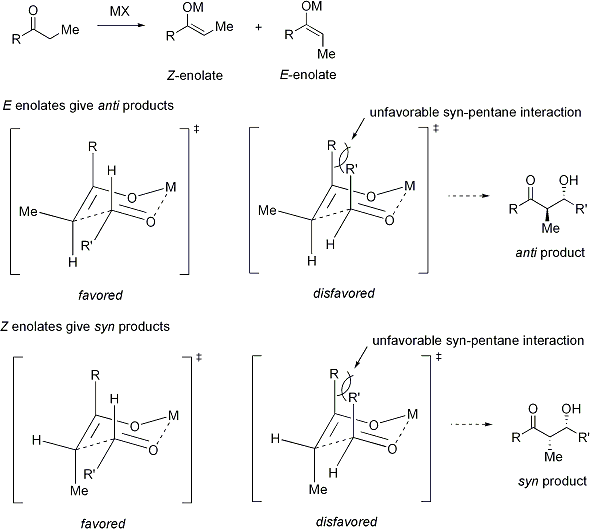
Contrôle dans la réaction Aldol
Le problème
Le problème de «contrôle» dans le addition aldolique est le mieux démontré par un exemple. Examiner les résultats de cette réaction hypothétique:
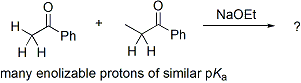
Dans cette réaction, deux cétones non symétriques sont condensés à l'aide l'éthylate de sodium. La basicité de l'éthylate de sodium est telle qu'elle ne peut déprotoner totalement ou l'autre des cétones, mais peut produire de petites quantités de l'énolate de sodium de deux cétones. En effet, cela signifie que en plus d'être électrophiles potentiels d'aldolisation, les deux cétones peuvent également agir en tant que nucléophiles via leur énolate de sodium. Deux électrophiles et nucléophiles deux puis des résultats potentiellement en quatre produits possibles:

Ainsi, si l'on souhaite obtenir un seul des produits croisés, alors on doit l'addition aldolique "de contrôle".
Acidité
Si un partenaire est beaucoup plus acide que l'autre, alors le contrôle peut être automatique. Le proton plus acide est prélevée par la base et un énolate est formé. Ce type de commande ne fonctionne que si la différence de l'acidité est suffisamment grande et pas d'excès de base est utilisée pour la réaction. Le contrôle est plus simple si seulement un des réactifs a protons acides et seulement cette molécule constitue l'énolate. Par exemple, l'addition de malonate de diéthyle dans benzaldéhyde serait seulement produire un produit:
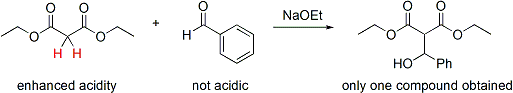
Dans ce cas, le doublement activé protons méthylène du malonate serait préférentiellement déprotoné par l'éthoxyde de sodium et former quantitativement l'énolate de sodium. Depuis benzaldéhyde n'a pas de protons alpha-acides, il est seulement possible une combinaison nucléophile-électrophile; par conséquent, le contrôle a été atteint. Notez que cette approche combine deux éléments de contrôle: augmentation de l'acidité des protons alpha sur le nucléophile et l'absence de protons alpha sur l'électrophile.
L'ordre d'addition
Une solution commune est de former l'énolate d'un partenaire de premier, puis ajouter l'autre partenaire dans contrôle cinétique. Signifie que le contrôle cinétique aldol réaction d'addition de l'avant doit être nettement plus rapide que la réaction rétro-aldol inverse. Pour que cette approche réussisse, deux autres conditions doivent aussi être remplies; savoir, il doit être possible de former quantitativement l'énolate d'un partenaire et la réaction aldolique l'avant doit être nettement plus rapide que le transfert de l'énolate d'un partenaire à l'autre. Des conditions de contrôle cinétique communes impliquent la formation de l'énolate d'une cétone avec LDA à -78 ° C, suivie par l'addition lente d'un aldéhyde.
Énolates
Formation
L'énolate peut être formée en utilisant une base forte («conditions difficiles») ou en utilisant un Acide de Lewis et d'une base faible («conditions douces»):
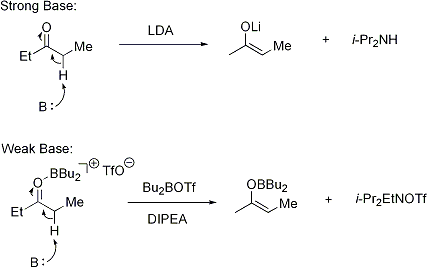
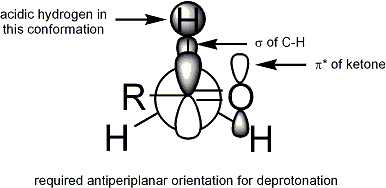
Pour déprotonation de se produire, la condition est que le stéréoélectronique alpha-CH liaison sigma doit être capable de se chevaucher avec la pi * orbital de la carbonyle:
Géométrie
Des études approfondies ont été réalisées sur la formation d'énolates dans diverses conditions. Il est maintenant possible de produire, dans la plupart des cas, la géométrie de l'énolate souhaité:
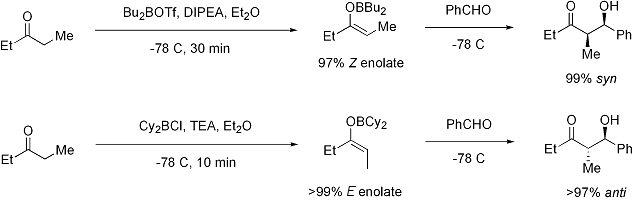
(- Dans l'image ci-dessus, le deuxième schéma de réaction devrait dire> 99% E-énolate, pas Z -) Pour les cétones, la plupart des conditions de énolisation donnent énolates Z. Pour esters, la plupart des conditions de énolisation donnent E énolates. L'addition de HMPA est connu pour inverser la stéréosélectivité de déprotonation.
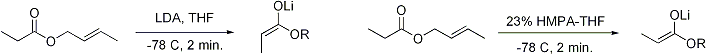
La formation stéréosélective des énolates a été rationalisée avec le soi-disant modèle Irlande, bien que sa validité est quelque peu discutable. Dans la plupart des cas, il ne est pas connue, le cas échéant, intermédiaires sont monomère ou oligomère dans la nature; néanmoins, le modèle Irlande reste un outil utile pour comprendre énolates.
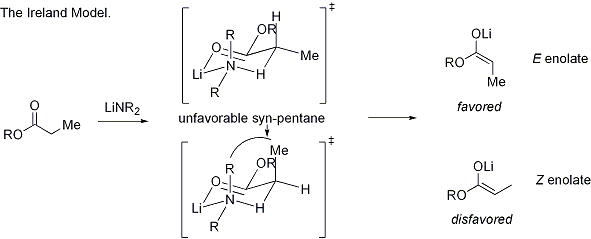
Dans le modèle Irlande, la déprotonation est supposé procéder par un état de transition monomère six chaînons. La plus grande des deux substituants sur l'électrophile (dans le cas ci-dessus, un groupe méthyle est supérieure à protons) adopte une disposition équatoriale dans l'état de transition favorisée, menant à une préférence pour E énolates. Le modèle ne est manifestement pas dans de nombreux cas; par exemple, si le mélange solvant est changé de THF à 23% HMPA-THF (comme on le voit ci-dessus), la géométrie de l'énolate est inexplicable inversée.
Vs Kinetic énolates thermodynamiques
Si une cétone asymétrique est soumis à une base, il a le potentiel de former des énolates deux régioisomères (en ignorant géométrie énolate). Par exemple:
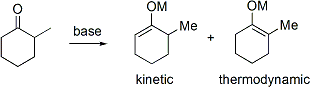
L'énolate trisubstitué est considéré comme le énolate cinétique tandis que l'énolate tétrasubstitué est considéré comme l'énolate thermodynamique. L'hydrogène alpha déprotoné pour former l'énolate cinétique est moins encombré, et donc déprotoné plus rapidement. En général, les oléfines tétrasubstituées sont plus stables que des oléfines trisubstituées en raison de la stabilisation hyperconjugaison. Le ratio de régioisomères énolates est fortement influencée par le choix de la base. Pour l'exemple ci-dessus, contrôle cinétique peut être établie avec LDA à -78 ° C, donnant 99: une sélectivité de cinétique: énolate thermodynamique, tandis que le contrôle thermodynamique peut être établie avec triphenylmethyllithium au la température ambiante, donnant 10:90 sélectivité.
En général, les énolates cinétiques sont favorisées par des températures relativement froides, des liaisons ioniques métal-oxygène, et la déprotonation rapide en utilisant un léger excès d'une base forte, tandis que les énolates entravé thermodynamiques sont favorisées par des températures plus élevées, des liaisons covalentes relativement métal-oxygène, et plus équilibration fois pour déprotonation en utilisant une petite quantité sous-stoechiométrique d'une base forte. Utilisation d'une quantité sous-stoechiométrique de base permet une petite fraction du composé carbonyle unenolized à équilibrer l'énolate thermodynamique au régioisomère en agissant comme une navette de protons.
Stéréosélectivité
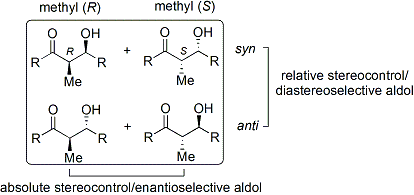
La réaction d'aldolisation est particulièrement utile parce que deux nouveaux centres stéréogènes sont générés dans une réaction. Des recherches approfondies ont été effectuées pour comprendre le mécanisme de réaction et d'améliorer la sélectivité observée dans de nombreuses conditions différentes. La convention de syn / anti est couramment utilisé pour désigner la stéréochimie relative à l'α- et β-carbone.
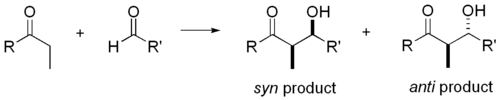
La convention se applique lorsque le propionate (ou ordre supérieur) nucléophiles sont ajoutés à aldéhydes. Le groupe R de la cétone et le groupe R 'de l'aldéhyde sont alignés selon un motif "zig zag" dans le plan du papier, et la disposition des stéréocentres formés est réputé syn ou anti, selon si elles sont sur le même ou côtés opposés de la chaîne principale.
Documents plus anciens utilisent le érythro - thréo nomenclature familière de la chimie des glucides.
E vs Z énolates
Il n'y a pas de différence significative entre le niveau de stereoinduction observé avec E et Z énolates:
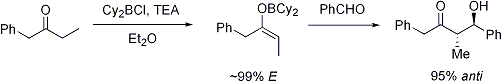
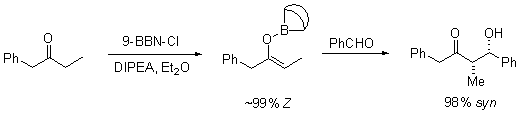
ion métallique
Le cation énolate de métal peut jouer un rôle important dans la détermination du niveau de stéréosélectivité dans la réaction d'aldol. bore est souvent utilisé parce que sa les longueurs de liaison sont sensiblement plus courte que celle des autres métaux tels que le lithium , l'aluminium ou le magnésium . Par exemple, les obligations de bore-carbone et de bore-oxygène sont 1.4 à 1.5 Å et 1.5 à 1.6 Å de longueur, respectivement, alors que le métal-carbone et des liaisons métal-oxygène typique sont généralement 1.9 à 2.2 Å et 2,0 à 2,2 Å de longueur, respectivement. Cela a pour effet de «resserrement» de la état de transition:
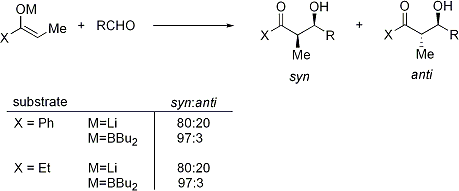
Stéréosélectivité: Alpha stéréocentre sur le énolate
La réaction d'aldolisation peut présenter "stéréocontrôle à base de substrat", dans laquelle existant chiralité de chaque réactif influe sur le résultat stéréochimique de la réaction. Cela a été largement étudié, et dans de nombreux cas, on peut prédire le sens de induction asymétrique, si ce ne est le niveau absolu de diastéréosélectivité. Si l'énolate contient stéréocentre en position alpha, une excellente stéréochimique peut être réalisée.
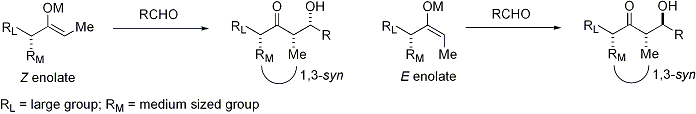
Dans le cas d'un énolate E, l'élément de commande est dominant allylique 1,3 souche alors que dans le cas d'un énolate Z, l'élément de commande dominante est d'éviter des interactions 1,3-diaxiales. Le modèle général est présenté ci-dessous:
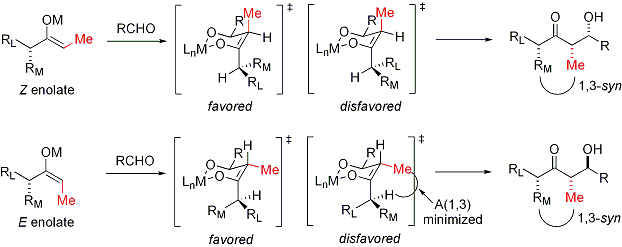
Pour plus de clarté, le stéréocentre sur l'énolate a été épimérisé; en réalité, le diastereoface opposé de l'aldéhyde aurait été attaqué. Dans les deux cas, le diastéréo-isomère 1,3-syn est favorisée. Il existe de nombreux exemples de ce type de stéréocontrôle:
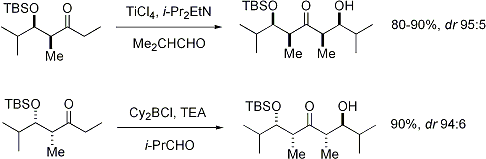
Stéréosélectivité: Alpha stéréocentre sur le électrophile
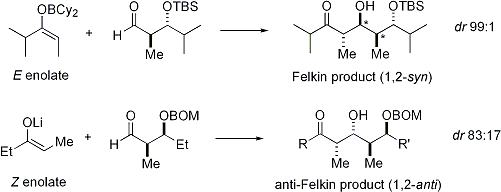
Lorsque énolates attaque aldéhydes avec un stéréocentre alpha, excellente stéréochimique est également possible. Le constat général est que E énolates exposition Felkin sélection de diastereoface, tandis que Z énolates présentent anti-Felkin sélectivité. Le modèle général est présenté ci-dessous:
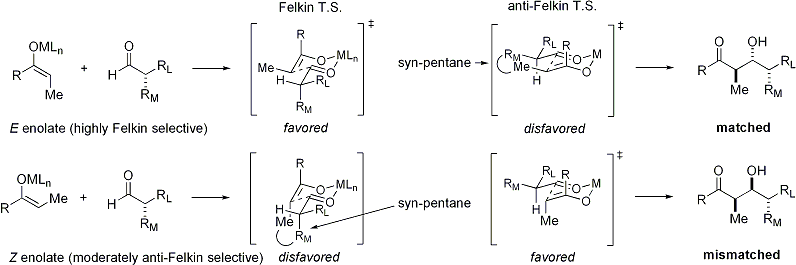
Depuis Z énolates doivent réagir à travers un état de transition qui contient soit une interaction syn-pentane déstabiliser ou anti-Felkin rotamère, Z-énolates présentent des niveaux inférieurs de diastéréosélectivité dans ce cas. Quelques exemples sont présentés ci-dessous:
Stéréosélectivité: modèle fusionné pour stereoinduction
Si à la fois l'énolate et la chiralité aldéhyde contiennent tous les deux pré-existante, alors le résultat de la «double stereodifferentiating" aldol réaction peut être prédit en utilisant un modèle stéréochimique fusionné qui prend en compte le biais énolate du visage, de la géométrie énolate, et aldéhyde biais du visage. Plusieurs exemples de l'application de ce modèle sont présentées ci-dessous: 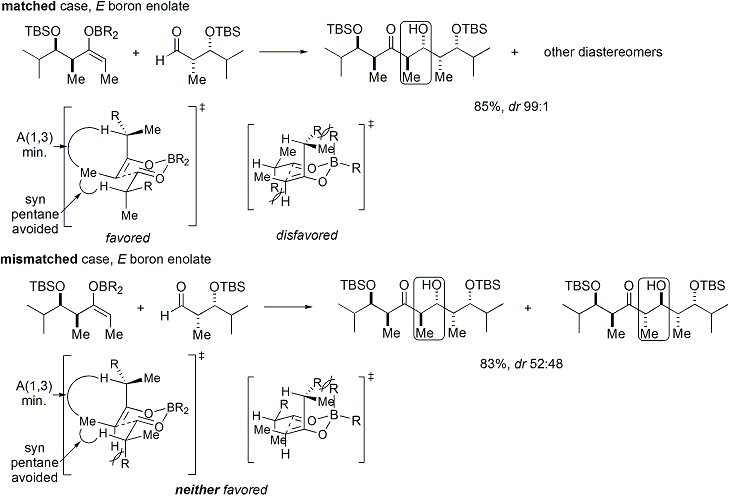
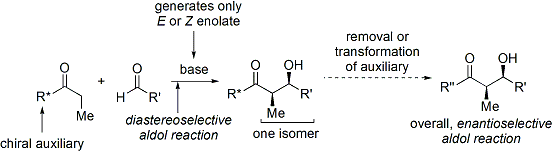
Oxazolidinone la chimie de Evans
Synthèses organiques modernes exigent maintenant la synthèse de composés dans forme énantiopur. Comme la réaction d'addition aldolique crée deux stéréocentres, jusqu'à quatre stéréoisomères peuvent en résulter.
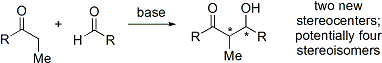
De nombreuses méthodes qui contrôlent à la fois la stéréochimie relative (c.-à-syn ou anti, comme discuté ci-dessus) et absolue stéréochimie (c.-à-R ou S) ont été développés.
Une méthode largement utilisée est le Evans ' acyl Procédé d'oxazolidinone. Développé dans les années 1970 et 1980 par le David A. Evans et ses collègues, la méthode fonctionne en créant temporairement un énolate chiral en ajoutant un auxiliaire chiral. La chiralité préexistante de l'auxiliaire est ensuite transféré dans le produit d'addition aldolique en effectuant une réaction d'aldolisation diastéréosélective. Lors de l'enlèvement ultérieur de l'auxiliaire, le stéréoisomère souhaité d'aldol est révélé.
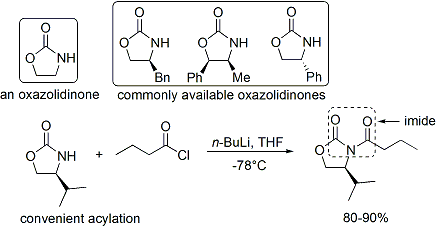
Dans le cas de la méthode de Evans, l'auxiliaire chiral est un annexée oxazolidinone, et le composé carbonylé est une résultante imide. Un certain nombre de oxazolidinones sont maintenant facilement disponibles dans les deux formes énantiomères. Ceux-ci peuvent coûter environ $ 10- $ 20 dollars par gramme, ce qui les rend relativement coûteux.
Le acylation d'une oxazolidinone est une procédure pratique et est officieusement appelé «chargement fait". Z-énolates, conduisant à des adduits syn-aldoliques, peuvent être formés de manière fiable à l'aide de bore médiation énolisation douce:
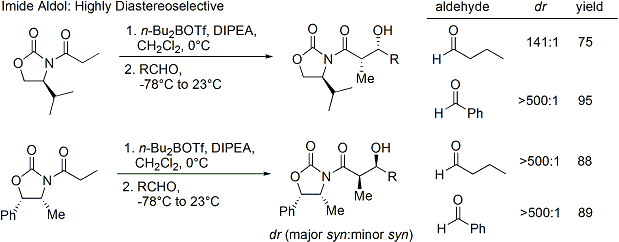
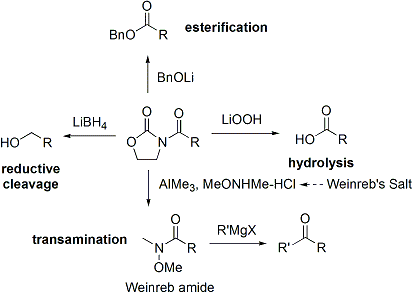
Souvent, un seul diastéréo-isomère peut être obtenu par une cristallisation du produit d'addition aldolique. Malheureusement, les produits d'addition anti-aldol ne peuvent être obtenues de manière fiable avec la méthode Evans. Malgré le coût et la limitation de ne donner que des produits d'addition syn, une fiabilité supérieure de la méthode, la facilité d'utilisation et la polyvalence rendre la méthode de choix dans de nombreuses situations. De nombreuses méthodes sont disponibles pour le clivage de l'auxiliaire:
Lors de la construction imide, à la fois des réactions d'addition aldolique syn et anti-sélectives peuvent être réalisées, ce qui permet l'assemblage de trois des quatre stereoarrays possibles: sélective syn: et anti sélective:
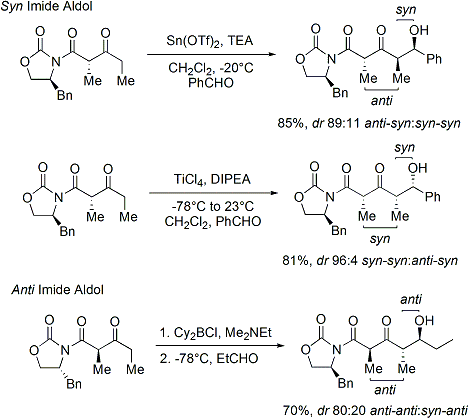
Dans les réactions de syn-sélective, les deux méthodes donnent énolisation de l'énolate de Z, comme prévu; cependant, le résultat stéréochimique de la réaction est contrôlée par le stéréocentre de méthyle, au lieu de la chiralité du oxazolidinone. Les méthodes décrites permettent l'assemblage stéréosélective polycétides, une classe de produits naturels qui présentent souvent le rétron aldolique.
Modern Chemistry Aldol
Méthodologie récente permet désormais une plus grande variété de réactions d'aldolisation à mener, souvent avec une quantité catalytique de ligand chiral. Lorsque les réactions utilisent de petites quantités de énantiomériquement purs ligands pour induire la formation de produits énantiomériquement purs, les réactions sont généralement appelés "catalytique asymétrique»; par exemple, un grand nombre catalytique différent, aldoliques réactions asymétriques sont maintenant disponibles.
Acétate de réactions d'aldolisation
Une limitation clé de la approche auxiliaire chiral décrit précédemment est l'échec de la N-acétyl imides de réagir de manière sélective. Une première approche est d'utiliser un temporaire groupe thioéther:
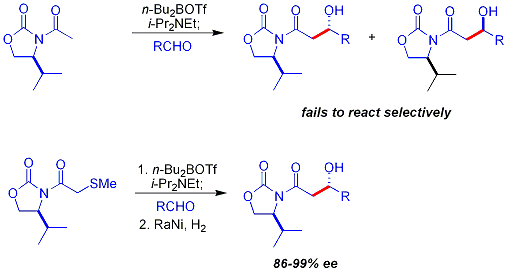
La réaction de Mukaiyama
Le La réaction de Mukaiyama est le addition nucléophile de les éthers de silyle à de aldéhydes catalysée par un Acide de Lewis tel que le trifluorure de bore ou le chlorure de titane. La réaction d'aldol Mukaiyama ne suit pas le modèle Zimmerman-Traxler. Carreira a décrit la méthodologie asymétrique particulièrement utile avec des acétals de cétènes silyle, remarquable pour ses niveaux élevés de énantiosélectivité et large champ de substrat.
La méthode fonctionne sur aldéhydes aliphatiques non ramifiés, qui sont souvent pauvres électrophiles, pour procédés catalytiques asymétriques. Cela peut être dû à la différenciation électronique et stérique pauvres entre leur enantiofaces.

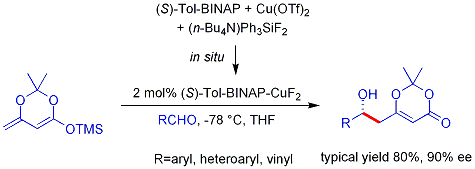
L'analogue vinylogue processus de aldol Mukaiyama peut également être rendue catalytique et asymétrique. L'exemple ci-dessous fonctionne efficacement pour les aldéhydes aromatiques (mais pas aliphatiques) et le mécanisme est censé impliquer, un diénolate de métal lié chiral.
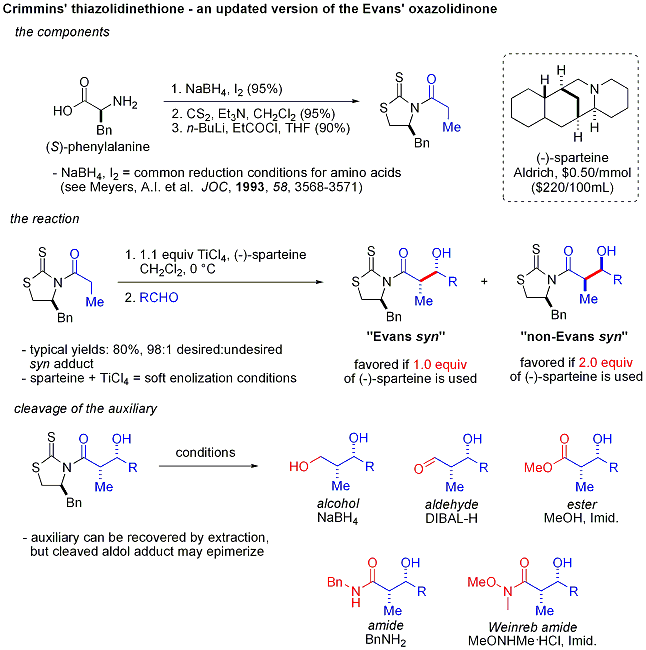
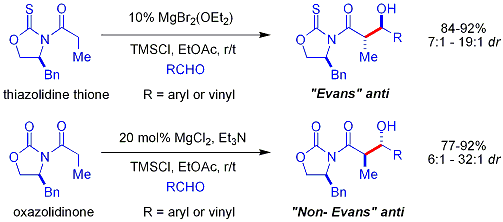
Crimmins thiazoldinethione aldol
Une version plus récente de l'auxiliaire du Evans est le thiazoldinethione Crimmins. Le rendements, diastéréosélectivités et énantiosélectivités de la réaction sont généralement élevés, bien que pas aussi élevé que dans les cas Evans comparables. Contrairement à l'auxiliaire Evans, cependant, le thiazoldinethione peut réaliser des réactions aldoliques acétate (ref:... Crimmins, Org Lett 2007, 9 (1), 149-152) et peut produire le «syn Evans" ou "non-Evans syn" des produits d'addition en faisant simplement varier la quantité de (-) - Spartéine. On pense que la réaction se déroule par l'intermédiaire de six chaînons, lié au titane états de transition, analogues aux états de transition proposées pour l'auxiliaire Evans.
Réactions aldoliques Organocatalytic
Un nouveau développement passionnant est l'utilisation de chiraux secondaires amine catalyseurs. Ces amines secondaires forment transitoire énamines lorsqu'ils sont exposés à des cétones, qui peuvent réagir avec des électrophiles énantiosélectivement aldéhydes appropriés. Ceci est connu comme la catalyse énamine, un type de organocatalyse, puisque le catalyseur est entièrement basé sur une petite molécule organique. Dans un exemple séminal, proline efficacement catalysée par la cyclisation d'un tricétone:
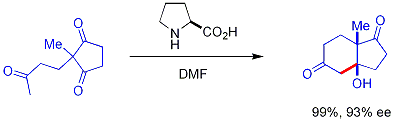
Cette réaction est connue comme la réaction Hajos-Parrish (aussi connu comme la réaction Hajos-Parrish-Eder-Sauer-Wiechert, se référant à un rapport de Schering contemporaine de la réaction dans des conditions plus sévères). Seule une quantité catalytique de proline est nécessaire (3% en mole). Il n'y a pas de danger d'une réaction de base achiral parce que les intermédiaires énamine transitoires sont beaucoup plus fortement nucléophiles que leurs énols de cétones parent. Cette stratégie est particulièrement puissante car elle offre un moyen simple de générer énantiosélectivité dans les réactions sans utiliser des métaux de transition, qui ont les inconvénients possibles d'être toxiques ou coûteux.
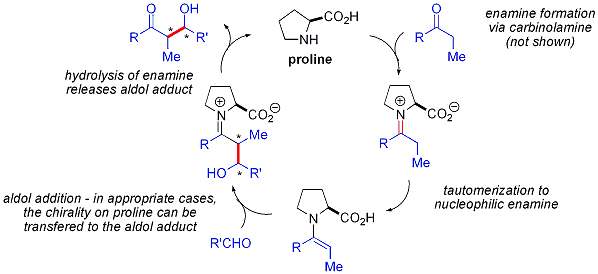
Fait intéressant, les réactions d'aldol catalysée par la proline ne montrent pas d'effets non linéaires (l'énantiosélectivité des produits est directement proportionnelle à la énantiopureté du catalyseur). Combiné avec preuves de marquage isotopique et des études informatiques , le projet de mécanisme de réaction pour les réactions d'aldol catalysée par la proline est la suivante:
Cette stratégie permet à la réaction croisée d'aldol autrement difficile entre deux aldéhydes. En général, les réactions croisées entre aldoliques aldéhydes sont généralement difficiles parce qu'ils ne peuvent polymériser facilement réagir ou non sélective pour obtenir un mélange statistique des produits. Le premier exemple est présenté ci-dessous:
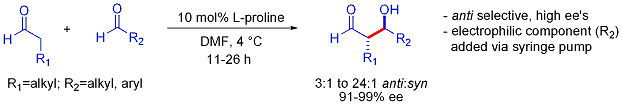
A la différence de la préférence pour des produits d'addition syn typiquement observés dans des ajouts à base d'aldolisation-énolates, ces ajouts d'aldolisation organocatalyzed sont anti-sélectif. Dans de nombreux cas, les conditions organocatalytic sont suffisamment douces pour éviter la polymérisation. Cependant, la sélectivité nécessite l'addition lente seringue-pompe commandée du partenaire électrophile souhaité car les deux partenaires ont généralement réagissant protons énolisables. Si un aldéhyde n'a pas de protons énolisables ou alpha ou bêta-ramification, un contrôle supplémentaire peut être atteint.
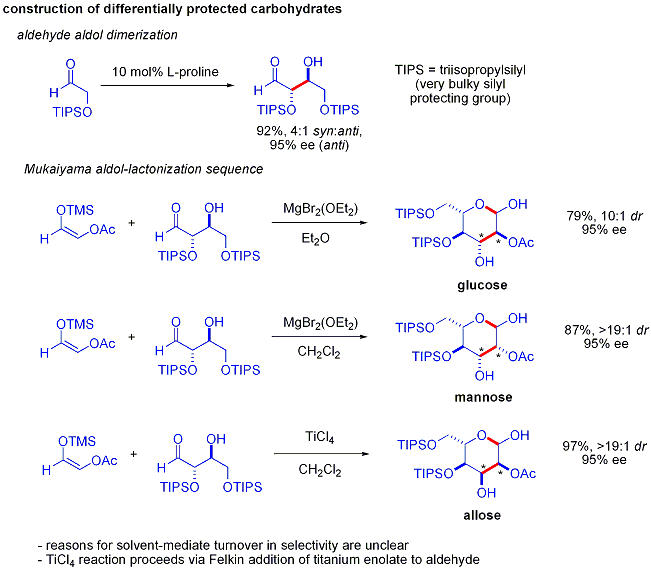
Une élégante démonstration de la puissance de réactions aldoliques organocatalytic asymétriques a été divulguée par MacMillan et ses collègues en 2004 dans leur synthèse des différentielle protégées glucides . Bien que les méthodes traditionnelles de synthèse accomplir la synthèse de hexoses aide de variantes du itérative les stratégies de protection-déprotection, ce qui nécessite 8 à 14 étapes, organocatalyse peuvent accéder à un grand nombre des mêmes substrats en utilisant un protocole efficace en deux étapes impliquant la dimérisation catalysée par de la proline d'alpha-oxyaldehydes suivie d'une cyclisation aldolique Mukaiyama tandem.
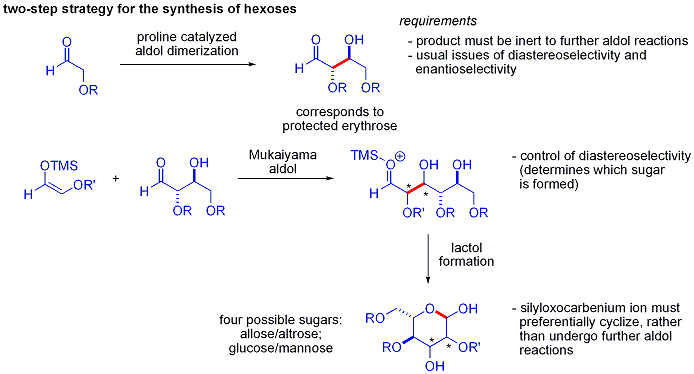
La dimérisation d'alpha-aldol oxyaldehydes exige que le produit d'addition d'aldol, un aldéhyde lui-même, soit inerte pour favoriser les réactions d'aldolisation. Des études antérieures ont révélé que palier d'aldéhydes alpha-alkyloxy ou alpha- silyloxy substituants sont approprié pour cette réaction, tandis que des aldéhydes portant les groupes attracteurs d'électrons tels que acétoxy étaient non réactif. La protégée produit d'érythrose pourrait ensuite être converti en sucres quatre possibles par addition d'aldol suivie par Mukaiyama formation lactol. Cela nécessite diastereocontrol approprié dans le addition aldolique Mukaiyama et le produit ions silyloxycarbenium à préférentiellement cyclisent, plutôt que de subir une autre réaction d'aldol. En fin de compte, le glucose , mannose, et allose ont été synthétisés:
additions "Direct" aldoliques
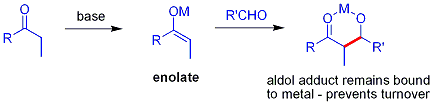
Dans l'addition aldolique d'habitude, un composé carbonyle est déprotoné pour former l'énolate. L'énolate est ajouté à un aldéhyde ou une cétone, qui forme un alcoxyde, qui est ensuite protoné sur bilan. Une méthode supérieure, en principe, permettrait d'éviter la séquence déprotonation-aldol-protonation en faveur d'une "addition aldolique directe". Le problème majeur dans un tel processus, ce est que l'addition aldolique génère un alcoolate, qui est beaucoup plus fondamental que les matières premières, ce qui exclut le chiffre d'affaires de catalyseur:
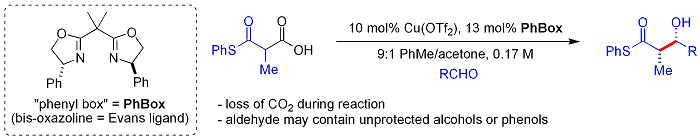
Une approche, récemment démontré par Evans, est de silyler le produit d'addition aldolique:
Cette méthode est plus rentable et industriellement utile que les procédures fondées énolates-plus typiques. Une approche plus récente, biomimétique par Shair utilise bêta-thioketoacids comme nucléophile. Le fragment est cétoacide décarboxylé in situ (le est un ligand chiral bisoxazoline). Fait intéressant, les aldéhydes aliphatiques et aromatiques ramifiés sont généralement de mauvais substrats.