
Hemofilia
Você sabia ...
Crianças SOS, uma instituição de caridade educação , organizou esta selecção. SOS mães cada um cuidar de uma família de crianças apadrinhadas .
| Hemofilia d | |
|---|---|
| Classificação e recursos externos | |
| CID- 10 | D 66- D 68 |
| CID- 9 | 286 |
| OMIM | 306700 306900 264900 |
| DiseasesDB | 5555 5561 29376 |
| MedlinePlus | 000537 |
| Medcenter | med / 3528 |
| MeSH | D025861 |
Hemofilia (também escrito como a hemofilia, a partir do grego haima "sangue" e philia "amar") é um grupo de hereditário doenças genéticas que comprometem a capacidade do corpo para controlar sangue coagulação ou coagulação. Na sua forma mais comum, A hemofilia A, a coagulação factor VIII está ausente. Em A hemofilia B, o factor IX é deficiente. A hemofilia A ocorre em cerca de 1 em 5.000-10.000 nascimentos do sexo masculino, enquanto Hemofilia B ocorre em cerca de 1 em cerca de 20,000-34,000.
Os efeitos desta ligada ao sexo, X desordem cromossômica manifestam-se quase inteiramente no sexo masculino, embora o gene para a doença é herdada da mãe. As fêmeas têm dois cromossomos X, enquanto os machos têm apenas um, na falta de um 'back-up' cópia do gene defeituoso. As fêmeas são, portanto, quase exclusivamente portadores da desordem, e pode ter herdado isso de qualquer mãe ou do pai. Em cerca de 30% dos casos de hemofilia B, no entanto, não existe história familiar da doença e a condição é o resultado de uma mutação no gene espontânea. Uma mãe que é uma transportador tem 50% de chance de passar o cromossomo X defeituoso a sua filha, enquanto um pai afetado será sempre passar o gene afetado para suas filhas. Um filho não podem herdar o gene defeituoso de seu pai.
Estas deficiências genéticas podem reduzir plasma sanguíneo níveis de fator de coagulação de fatores de coagulação necessários para um processo de coagulação normais. Quando um vaso sanguíneo é lesado, uma crosta temporária faz formar, mas os factores de coagulação em falta evitar a formação de fibrina, que é necessário para manter o coágulo de sangue. Assim, um hemofílico não sangra mais intensamente do que uma pessoa normal, mas durante muito mais tempo. Em hemofílicos graves, mesmo uma pequena lesão pode resultar em perda de sangue duradouro dias, semanas, ou nunca curar completamente. O risco crítico aqui é normalmente com pequenas lesões que, devido ao fator VIII faltando, tomar longos tempos de curar. Em áreas como o cérebro ou dentro articulações isso pode ser fatal ou permanentemente debilitantes.
O sangramento com externo lesão é normal, mas a incidência de tarde re-sangramento e hemorragia interna é aumentada, especialmente em músculos, articulações, ou sangramento em espaços fechados. As principais complicações incluem hemartrose, hemorragia , hemorragia gastrointestinal, e menorragia.
Causas
A hemofilia é quase sempre causada por um defeito genético que causa a falta de um factor de coagulação do funcionamento normal:
- A hemofilia A envolve uma falta de coagulação funcional Factor VIII. (Isto representa 90% dos casos de hemofilia).
- A hemofilia B envolve uma falta de coagulação funcional Fator IX.
- A hemofilia C envolve uma falta de coagulação funcional Fator XI.
- Hipofibrinogenemia envolve uma falta de coagulação Fator funcional I. Porque é tão rara, cerca de 1 a 2 casos por milhão de nascimentos, não tem tratamento definitivo aprovado pelo FDA. Afeta homens e mulheres igualmente. O sangue de pessoas com hipofibrinogenemia nem coágulos nem contém quantidades suficientes de Fibrinogênio.
Ocorrência
A hemofilia é bastante raro, com apenas cerca de 1 instância em cada 10.000 nascimentos (ou 1 em cada 5.000 nascimentos do sexo masculino) para a hemofilia A e 1 em 50.000 nascimentos para hemofilia B. Cerca de 18.000 pessoas nos Estados Unidos têm hemofilia. A cada ano em os EUA, cerca de 400 bebês nascem com a doença. Hemofilia geralmente ocorre em homens e menos frequentemente no sexo feminino. Estima-se que cerca de 2500 canadenses têm hemofilia A e cerca de 500 canadenses têm hemofilia B.
História
A mais antiga referência implícita possível hemofilia pode ter sido no Talmud, um judeu texto sagrado, que afirma que os homens não têm de ser circuncidado se dois irmãos já tinham morrido do procedimento. Em 1000, o Médico árabe Abu al-Qasim al-Zahrawi (conhecido como Albucasis no Ocidente) escreveu uma descrição mais explícita da hemofilia em sua Al-Tasrif, no qual ele escreveu sobre uma Família andaluza cujos machos morreu de hemorragia após ferimentos leves.
Em 1803 , o Dr. John Conrad Otto, um médico da Filadélfia, escreveu um relato sobre "uma disposição hemorrágica existente em certas famílias." Ele reconheceu que a doença era hereditária e que isso afetou machos e fêmeas raramente. Ele foi capaz de rastrear a doença de volta para uma mulher que se estabeleceu perto de Plymouth em 1720. O primeiro uso do termo "hemofilia" aparece em uma descrição da condição escrito por Hopff na Universidade de Zurique em 1828 . Em 1937 , Patek e Taylor, dois médicos de Harvard, descobriu globulina anti-hemofílico. Pavlosky, um médico de Buenos Aires, encontrados A hemofilia A e A hemofilia B para ser doenças separadas, fazendo um teste de laboratório. Este teste foi feito por transferência do sangue de um hemofílico para outro hemofílico. O facto de este corrigido o problema de coagulação mostrou que havia mais de uma forma de hemofilia.
Hemofilia em realeza europeia destaque e, assim, às vezes é conhecido como "a doença real". Rainha Victoria passou a mutação de seu filho Leopold e, através de várias das suas filhas, a vários membros da realeza de todo o continente, incluindo as famílias reais da Espanha, Alemanha, e Rússia. Alexei Nikolaevich, filho de Nicholas II, era um descendente da rainha Victoria e sofria de hemofilia. Alegou-se que Rasputin foi bem-sucedida no tratamento da Alexei da hemofilia da Rússia. No momento, um tratamento comum administrados por profissionais médicos foi usar a aspirina, o que piorou em vez de diminuir o problema. Acredita-se que, simplesmente pelo desaconselha o tratamento médico, Rasputin podem trazer uma melhora significativa visível e à condição de Alexei.
Antes de 1985, não havia leis promulgadas dentro os EUA para rastrear sangue. Como resultado, muitos pacientes hemofílicos que receberam fator de coagulação não testados e sem tela antes de 1992 estavam em um risco extremo de contrair HIV e A hepatite C através destes produtos sanguíneos. Estima-se que mais de 50% da população Hemofilia, mais de 10.000 pessoas, contraíram o HIV a partir do fornecimento de sangue contaminado nos Estados Unidos sozinho.
Como resultado direto de a contaminação do suprimento de sangue no final de 1970 e início de meados de 1980 / com vírus como o Hepatite e do HIV , foram desenvolvidos novos métodos para a produção de produtos de factor de coagulação. A resposta inicial foi para aquecer-treat ( pasteurizar) Factor concentrado derivado de plasma, seguido pelo desenvolvimento do factor monoclonal concentrados, que utilizam uma combinação de tratamento de calor e cromatografia de afinidade, para inactivar quaisquer agentes virais no plasma reunido a partir do qual o concentrado de factor é derivado. O Lindsay tribunal na Irlanda investigado, entre outras coisas, a aprovação lenta dos novos métodos.
Genética
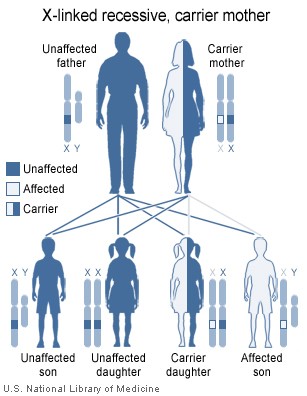
As fêmeas possuem dois cromossomos X, enquanto os machos têm um X e um Cromossomo Y. Uma vez que as mutações que provocam a doença são recessiva, uma mulher que carregava o defeito em um de seus cromossomos X não pode ser afetado por ela, como o equivalente alelo em seu outro cromossomo deve expressar-se para produzir os fatores de coagulação necessários. No entanto, o cromossomo Y em homens não tem gene para fatores VIII ou IX. Se os genes responsáveis pela produção de o factor VIII ou fator IX presente no cromossomo X de um macho são deficientes não há nenhum equivalente no cromossomo Y, de modo que o gene deficiente não é mascarado pelo alelo dominante e ele irá desenvolver a doença.
Uma vez que um homem recebe seu único cromossomo X de sua mãe, o filho de uma mulher saudável silenciosamente portador do gene deficiente terá uma chance de 50% de herdar esse gene dela e com ela a doença; e se sua mãe é afetada com hemofilia, ele terá uma chance de 100% de ser um hemofílico. Em contraste, para uma mulher para herdar a doença, ela deve receber duas deficientes X-cromossomas, um da mãe e outro do pai (que deve, portanto, ser ele mesmo um hemofílico). Daí hemofilia é muito mais comum entre homens do que mulheres. No entanto, é possível que mulheres portadoras de se tornar hemofílicos amenas devido à lyonisation (inativação) dos cromossomos X. Filhas hemofílicos são mais comuns do que eram antes, como melhores tratamentos para a doença têm permitido mais hemofílico machos para sobreviver até a idade adulta e se tornam pais. Fêmeas adultas podem experimentar menorragia (períodos pesados), devido à tendência de sangramento. O padrão de herança é do tipo cruzado. Esse tipo de padrão também é visto em daltonismo .
Tal como acontece com todas as desordens genéticas, é, claro, também possível que um humano adquirir espontaneamente através mutação, em vez de herdar, por causa de uma nova mutação em um dos gâmetas dos seus pais. As mutações espontâneas são responsáveis por cerca de 33% de todos os hemofilia A e 20% de todos hemofilia B casos. Os testes genéticos e aconselhamento genético é recomendado para famílias com hemofilia. Teste pré-natal, como o amniocentese, está disponível para as mulheres grávidas que podem ser portadores da doença.
Probabilidade
Se uma fêmea dá à luz um filho hemofílico, ou a fêmea é um portador da doença ou a hemofilia foi o resultado de um mutação espontânea. Até direta moderno Testes de DNA, no entanto, não foi possível determinar se uma mulher com apenas crianças saudáveis foi um transportador ou não. Geralmente, os filhos mais saudáveis ela deu à luz, maior a probabilidade de que ela não era portadora. Se o Factor de RH do macho nascido é diferente da mãe, a criança não irá ser afectada.
Se um macho é afligido com a doença e tem filhos, suas filhas serão portadoras de hemofilia. Seus filhos, porém, não serão afetados com a doença. Isso ocorre porque a doença é ligada ao X eo pai não pode passar hemofilia através do cromossomo Y. Homens com o transtorno são, então, não mais propensos a passar sobre o gene para seus filhos do que mulheres portadoras, embora todos eles procriar filha vai ser portadores e todos os filhos que o pai não têm hemofilia (a menos que a mãe é portadora).
Tratamento
Embora não há cura para a hemofilia, pode ser controlada com infusões periódicas do factor de coagulação deficiente, isto é, o factor VIII na hemofilia A ou factor IX na hemofilia B. substituição do Factor pode ser isolado a partir de humano soro sanguíneo, recombinante, ou uma combinação dos dois. Alguns hemofílicos desenvolvem anticorpos (inibidores) contra os factores de substituição que lhes são atribuídos, de modo que a quantidade do factor tem de ser aumentada ou os produtos de substituição não-humanos deve ser dada, tal como o factor VIII porcino.
Se um paciente se torna refractário ao factor de coagulação de substituição, como resultado de inibidores que circulam, este pode ser parcialmente ultrapassada com humano recombinante fator VII (NovoSeven), que está registrado para esta indicação em muitos países.
No início de 2008, os EUA Food and Drug Administration aprovou Xyntha ( Wyeth) factor anti-hemofílico, geneticamente modificada a partir dos genes de células de ovário de hamster chinês. Desde 1993 (Dr. Mary Nugent) produtos de fator recombinante (que são normalmente cultivadas em ovário de hamster chinês ( CHO) células de cultura de tecidos e envolvem pouco, se algum produtos de plasma humano) estão disponíveis e têm sido amplamente utilizados em países ocidentais ricos. Enquanto os produtos de fator de coagulação recombinante oferecer maior pureza e segurança, são, como concentrado, extremamente caros, e geralmente não disponíveis no mundo em desenvolvimento. Em muitos casos, os produtos de fator de qualquer tipo são difíceis de obter em países em desenvolvimento.
Nos países ocidentais, as normas comuns de cuidados de cair em uma das duas categorias: profilaxia ou sob demanda. Profilaxia envolve a infusão de fator de coagulação em uma programação regular, a fim de manter os níveis de coagulação suficientemente elevada para impedir episódios de sangramento espontâneos. On-demand tratamento envolve o tratamento de episódios hemorrágicos, uma vez que eles surjam. Em 2007, um ensaio clínico foi publicada no New England Journal of Medicine (NEJM) comparando o tratamento on-demand de meninos (<30 meses) com hemofilia A com tratamento profilático (infusões de 25 UI / kg de peso corporal Fator VIII a cada dois dias) em relação ao seu efeito sobre a prevenção de doenças articulares-. Quando os rapazes atingiu 6 anos de idade, 93% daqueles no grupo profilaxia e 55% daqueles no grupo episódica-terapia teve um índice normal junta-estrutura sobre MRI. O tratamento profilático, contudo, resultou em média custos de $ 300.000 por ano. O autor de um editorial publicado na mesma edição do NEJM exige mais estudos clínicos que abordam a relação custo-eficácia do tratamento profilático.
Recomenda-se que as pessoas afectadas com hemofilia fazer exercícios específicos para fortalecer as articulações, principalmente nos cotovelos, joelhos e tornozelos. Os exercícios incluem elementos que aumentam a flexibilidade, o tom ea força dos músculos, aumentando a sua capacidade de proteger as articulações de sangramentos prejudiciais. Estes exercícios são recomendados após uma hemorragia interna e ocorre em uma base diária para fortalecer os músculos e articulações para evitar novos problemas de sangramento. Muitos exercícios recomendados incluem esportes padrão de aquecimento e exercícios de treinamento, como alongamento dos bezerros, círculos de tornozelo, flexão de cotovelo, e conjuntos quadríceps.
Os tratamentos alternativos e complementares
Estudos científicos indicam que a hipnose ea auto-hipnose pode ser eficaz na redução de sangramentos ea gravidade da sangramentos e assim a frequência do tratamento fator. Ervas que fortalecem os vasos sanguíneos e agir como adstringentes também podem beneficiar pacientes com hemofilia. Estas ervas incluem: Boldo ( Vaccinium myrtillus), extrato de semente de uva ( Vitis vinifera), Scotch vassoura ( Cytisus scoparius), Urtiga ( Urtica dioica), Hamamélis ( Hamamelis virginiana), e Yarrow ( Achillea millefolium).
O diagnóstico diferencial
A hemofilia A pode ser mimetizado pela Doença de von Willebrand
- Doença de von Willebrand tipo 2A, onde a diminuição dos níveis de factor de von Willebrand pode levar à proteólise prematura do Factor VIII. Em contraste com hemofilia, vWD tipo 2A é herdada de forma autossômica dominante.
- Doença de von Willebrand tipo 2N, onde Fator de von Willebrand não pode vincular Fator VIII, herança autossômica recessiva. (Ou seja, os pais devem dar à criança uma cópia do gene).
- Doença de von Willebrand Tipo 3, em que a ausência de Factor de von Willebrand provoca a proteólise prematura do Factor VIII. Em contraste com hemofilia, vWD tipo 3 é herdada de forma autossómica recessiva.