

Reacción en cadena de la polimerasa
Sabías ...
SOS cree que la educación da una mejor oportunidad en la vida para los niños en el mundo en desarrollo también. Madres SOS cada aspecto después de un una familia de niños apadrinados .

La reacción en cadena de la polimerasa (PCR) es una técnica ampliamente utilizada en biología molecular. Deriva su nombre de uno de sus componentes clave, un ADN polimerasa utilizados para amplificar un fragmento de ADN por in vitro enzimática replicación. A medida que avanza la PCR, el ADN así generado es en sí mismo utilizado como una plantilla para la replicación. Esto pone en marcha una reacción en cadena en la que el molde de ADN se exponencialmente amplificado. Con la PCR es posible amplificar una sola o pocas copias de un trozo de ADN a través de varios órdenes de magnitud, millones de generación o más copias de la pieza de ADN. PCR puede modificarse ampliamente para realizar una amplia gama de manipulaciones genéticas.
Casi todas las aplicaciones de la PCR emplean una ADN polimerasa estable al calor, tales como Taq polimerasa, una enzima originalmente aislado de la bacteria Thermus aquaticus. Este ADN polimerasa enzimáticamente ensambla una nueva cadena de ADN a partir de bloques de construcción de ADN, la nucleótidos, utilizando ADN de cadena sencilla como molde y Oligonucleótidos de ADN (también llamados Cebadores de ADN) necesarios para la iniciación de la síntesis de ADN. La gran mayoría de los métodos de PCR uso el ciclado térmico, es decir, alternativamente el calentamiento y el enfriamiento de la muestra de PCR a una serie definida de pasos de temperatura. Estos pasos de ciclos térmicos son necesarias para separar físicamente las hebras (a altas temperaturas) en un doble hélice de ADN ( Fusión del ADN), utilizado como plantilla durante la síntesis de ADN (a temperaturas más bajas) por la ADN polimerasa para amplificar selectivamente el ADN diana. La selectividad de PCR resulta de la utilización de cebadores que son complementario a la región de ADN específica para la amplificación en condiciones de ciclos térmicos específicos.
Desarrollado en 1983 por Kary Mullis, PCR es ahora una técnica común y, a menudo indispensable utilizado en los laboratorios de investigación médica y biológica para una variedad de aplicaciones. Éstos incluyen La clonación de ADN para basado en el ADN secuenciación, filogenia, o el análisis funcional de genes; el diagnóstico de enfermedades hereditarias; la identificación de huellas genéticas (utilizados en ciencias forenses y pruebas de paternidad); y la detección y el diagnóstico de enfermedades infecciosas . En 1993 Mullis ganó el Premio Nobel de Química por su trabajo en la PCR.
Principios y el procedimiento de PCR


PCR se utiliza para amplificar regiones específicas de una cadena de ADN (ADN diana). Esto puede ser un solo gen, una parte de un gen, o una secuencia no codificante. La mayoría de los métodos de PCR típicamente amplifican fragmentos de ADN de hasta 10 kilo pares de bases (kb), aunque algunas técnicas permiten la amplificación de fragmentos de hasta 40 kb de tamaño.
Una PCR configuración básica requiere varios componentes y reactivos. Estos componentes incluyen:
- Molde de ADN que contiene la región de ADN (objetivo) a amplificar.
- Dos cebadores, que son complementaria a las regiones de ADN en el 5 '(prime cinco) o 3 '(prime tres) extremos de la región de ADN.
- La ADN polimerasa tal como Taq polimerasa u otra ADN polimerasa con un óptimo de temperatura a alrededor de 70 ° C.
- Desoxinucleósidos trifosfato (dNTPs; también muy llamados comúnmente y erróneamente desoxinucleótidos trifosfato), los bloques de construcción de la cual las polimerasas de ADN sintetiza una nueva hebra de ADN.
- Solución tampón, proporcionando un ambiente químico adecuado para la actividad y la estabilidad de la ADN polimerasa óptima.
- Divalentes cationes , magnesio o manganeso iones; generalmente Mg 2+ se utiliza, pero Mn 2+ puede ser utilizado para la mutagénesis de ADN mediada por PCR, como una mayor concentración de Mn 2+ aumenta la tasa de error durante la síntesis de DNA
- Catión monovalente de potasio iones.
La PCR se lleva a cabo habitualmente en un volumen de reacción de 10-200 l en tubos de reacción pequeños (0,2-0,5 ml volúmenes) en una termociclador. El ciclador térmico se calienta y enfría los tubos de reacción para alcanzar las temperaturas requeridas en cada paso de la reacción (véase más adelante). Muchos termocicladores modernos hacen uso de la Efecto Peltier que permite tanto la calefacción y la refrigeración del bloque que sostiene los tubos de PCR simplemente invirtiendo la corriente eléctrica. Tubos de reacción de paredes finas permiten conductividad térmica favorable para permitir un rápido equilibrio térmico. La mayoría de los termocicladores tienen tapas calientes para evitar la condensación en la parte superior del tubo de reacción. Termocicladores mayores que carecen de una tapa calentada requieren una capa de aceite en la parte superior de la mezcla de reacción o una bola de cera en el interior del tubo.
Procedimiento
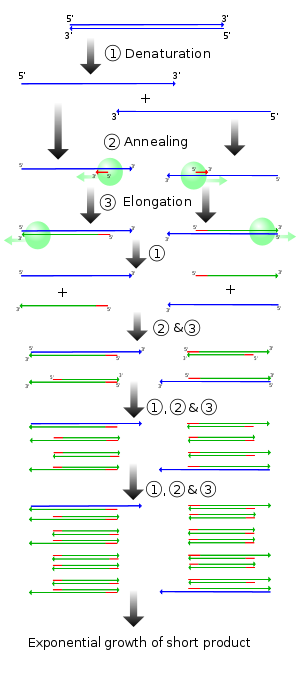
La PCR normalmente consiste en una serie de 20 a 40 cambios de temperatura repetidos llamados ciclos; cada ciclo consiste típicamente en 2-3 pasos discretos de temperatura. Más comúnmente PCR se lleva a cabo con los ciclos que tienen tres pasos de temperatura (Fig. 2). El ciclismo es a menudo precedida por un solo paso de temperatura (llamada retenida) a una temperatura elevada (> 90 ° C), y seguida por una espera al final de la extensión del producto final o breve almacenamiento. Las temperaturas utilizadas y la longitud de tiempo que se aplican en cada ciclo dependen de una variedad de parámetros. Estos incluyen la enzima utilizada para la síntesis de ADN, la concentración de iones divalentes y dNTPs en la reacción, y la temperatura de fusión ( Tm) de los cebadores.
- Etapa de inicialización: Esta etapa consiste en calentar la reacción a una temperatura de 94-96 ° C (o 98 ° C si se utilizan polimerasas extremadamente termoestables), que se mantiene durante 1-9 minutos. Sólo se requiere para ADN polimerasas que requieren la activación por calor por PCR de arranque en caliente .
- Etapa de desnaturalización: Este paso es el primer evento regular ciclismo y consiste en calentar la reacción a 94-98 ° C durante 20-30 segundos. Causa de fusión de plantilla de ADN y los cebadores mediante la interrupción de los enlaces de hidrógeno entre las bases complementarias de las hebras de ADN, produciendo cadenas simples de ADN.
- Etapa de recocido: La temperatura de reacción baja a 50-65 ° C durante 20-40 segundos que permiten la hibridación de los cebadores a la plantilla de ADN de cadena sencilla. Típicamente, la temperatura de recocido es de aproximadamente 3-5 grados Celsius por debajo de la Tm de los cebadores utilizados. Enlaces de hidrógeno ADN-ADN estable sólo se forman cuando la secuencia del cebador coincide muy de cerca la secuencia molde. La polimerasa se une al híbrido cebador-molde y comienza la síntesis de ADN.
- Etapa de extensión / alargamiento: La temperatura en este paso depende de la ADN polimerasa utilizada; Taq polimerasa tiene su óptima temperatura de la actividad a 75-80 ° C, y comúnmente una temperatura de 72 ° C se utiliza con esta enzima. En este paso la ADN polimerasa sintetiza una nueva hebra de ADN complementaria a la cadena molde de ADN mediante la adición de dNTPs que son complementarios a la plantilla en 5 'a 3', 5'-condensar el grupo fosfato de los dNTPs con la 3'- grupo hidroxilo en el extremo de la naciente (se extiende) cadena de ADN. El tiempo de extensión depende tanto de la ADN polimerasa utilizada y de la longitud del fragmento de ADN a amplificar. Como regla de oro-en su temperatura óptima, la ADN polimerasa polimeriza mil bases por minuto. En condiciones óptimas, es decir, si no hay limitaciones debido a la limitación de sustratos o reactivos, en cada etapa de extensión, se duplica la cantidad de ADN diana, dando lugar a exponencial (geométrica) la amplificación del fragmento de ADN específico.
- Elongación final: Este solo paso de vez en cuando se realiza a una temperatura de 70-74 ° C durante 5-15 minutos después del último ciclo de PCR para asegurar que cualquier ADN monocatenario restante está totalmente extendido.
- Asimiento final: Este paso a 4-15 ° C por tiempo indefinido se puede emplear para el almacenamiento a corto plazo de la reacción.

Para comprobar si la PCR generado el fragmento de ADN esperado (también denominado a veces como el amplímero o amplicón), electroforesis en gel de agarosa se emplea para la separación de tamaño de los productos de PCR. El tamaño (s) de productos de PCR se determina por comparación con una escalera de ADN (un marcador de peso molecular), que contiene fragmentos de ADN de tamaño conocido, se ejecutan en el gel junto a los productos de la PCR (ver Fig. 3).
Etapas de PCR
El proceso de PCR se puede dividir en tres etapas:
Amplificación exponencial: en cada ciclo, se duplica la cantidad de producto (suponiendo 100% de eficiencia de reacción). La reacción es muy específica y precisa.
Nivelación del escenario: La reacción se ralentiza como la ADN polimerasa pierde actividad y el consumo de reactivos, tales como los dNTP y cebadores hace que éstas se limitante.
Meseta: No más producto se acumula debido al agotamiento de los reactivos y enzimas.
Optimización de PCR
En la práctica, la PCR puede fallar por diversas razones, en parte debido a su sensibilidad a la contaminación que causa la amplificación del ADN productos espurios. Debido a esto, una serie de técnicas y procedimientos se han desarrollado para la optimización de las condiciones de PCR. La contaminación con ADN extraño se aborda con los protocolos y procedimientos que separan mezclas pre-PCR de contaminantes potenciales de ADN de laboratorio. Esto implica generalmente la separación espacial de las zonas PCR-configuración de las áreas de análisis o purificación de productos de PCR, y la limpieza minuciosa de la superficie de trabajo entre las configuraciones de reacción. Primer-técnicas de diseño son importantes para mejorar el rendimiento del producto PCR y evitar la formación de productos espurios, y el uso de los componentes del tampón alternos o enzimas polimerasas pueden ayudar con la amplificación de las regiones largas o de otro modo problemáticos de ADN.
Aplicación de PCR
Aislamiento de ADN genómico
PCR permite el aislamiento de fragmentos de ADN a partir de ADN genómico mediante amplificación selectiva de una región específica de ADN. Este uso de la PCR aumenta muchos métodos, tales como la generación sondas de hibridación para Southern o norte de la hibridación y Clonación de ADN, que requieren cantidades más grandes de ADN, lo que representa una región de ADN específica. PCR suministra estas técnicas con altas cantidades de ADN puro, lo que permite el análisis de muestras de ADN incluso de cantidades muy pequeñas de material de partida.
Otras aplicaciones de PCR incluyen La secuenciación del ADN para determinar las secuencias de PCR amplificado desconocidos en el que uno de los cebadores de amplificación pueden utilizarse en la secuenciación de Sanger, el aislamiento de una secuencia de ADN para acelerar tecnologías de ADN recombinante que implican la inserción de una secuencia de ADN en una plásmido o el material genético de otro organismo. Las colonias bacterianas ( E. coli) se puede cribar rápidamente por PCR de ADN correcta construcciones de vectores. PCR también se puede utilizar para huella genética; una técnica forense utiliza para identificar a una persona u organismo mediante la comparación de ADN experimentales a través de diferentes métodos basados en PCR.
Métodos 'huellas dactilares' Algunos de PCR tienen un alto poder de discriminación y pueden ser utilizados para identificar las relaciones genéticas entre los individuos, como entre padres e hijos o entre hermanos, y se utilizan en las pruebas de paternidad (Fig. 4). Esta técnica también se puede utilizar para determinar las relaciones evolutivas entre organismos.

La amplificación y cuantificación de ADN
Debido a que la PCR amplifica las regiones de ADN que se dirige, la PCR se puede utilizar para analizar cantidades muy pequeñas de muestra. Esto es a menudo crítico para análisis forense, cuando sólo una cantidad traza de ADN está disponible como evidencia. PCR también se puede utilizar en el análisis de ADN antiguo que tiene miles de años de edad. Estas técnicas basadas en PCR se han utilizado con éxito en animales, tales como cuarenta mil años de mamut , y también en el ADN humano, en aplicaciones que van desde el análisis de los egipcios momias a la identificación de un ruso Zar.
Métodos de PCR cuantitativos permiten la estimación de la cantidad de una determinada secuencia presente en una muestra - una técnica a menudo se aplica para determinar cuantitativamente los niveles de la expresion genica. PCR en tiempo real es una herramienta establecida para la cuantificación de ADN que mide la acumulación de producto de ADN después de cada ronda de amplificación por PCR.
- Ver también El uso de ADN en la entomología forense
PCR en el diagnóstico de enfermedades
PCR permite el diagnóstico precoz de enfermedades malignas tales como leucemia y linfomas, que es actualmente el más alto desarrollado en la investigación del cáncer, y ya se está utilizando de forma rutinaria. Los ensayos de PCR se pueden realizar directamente en muestras de ADN genómico para detectar células malignas-translocación específica a una sensibilidad que es al menos 10.000 veces más alta que otros métodos.
PCR también permite la identificación de microorganismos no cultivables o de crecimiento lento tales como micobacterias, bacterias anaerobias, o virus de los ensayos de cultivo de tejidos y modelos animales. La base para aplicaciones de diagnóstico de PCR en microbiología es la detección de agentes infecciosos y la discriminación de los no patógenos a partir de cepas patógenas en virtud de genes específicos.
Viral DNA igualmente puede ser detectado por PCR. Los cebadores utilizados deben ser específicos a las secuencias específicas en el ADN de un virus, y la PCR se pueden utilizar para los análisis de diagnóstico o la secuenciación de ADN del genoma viral. La alta sensibilidad de la PCR permite la detección de virus pronto después de la infección e incluso antes de la aparición de la enfermedad. Dicha detección temprana puede dar a los médicos una ventaja significativa en el tratamiento. La cantidad de virus ("carga viral") en un paciente también puede ser cuantificado mediante técnicas de cuantificación de ADN basados en la PCR (véase más adelante).
Las variaciones en la técnica básica de PCR
- PCR específica de alelo: Esta técnica de diagnóstico o de clonación se utiliza para identificar o utilizar polimorfismos de un solo nucleótido (SNP) (diferencias de una sola base en el ADN). Se requiere conocimiento previo de una secuencia de ADN, incluidas las diferencias entre alelos, y usa cebadores cuyos extremos 3 'abarcar el SNP. La amplificación por PCR en condiciones rigurosas es mucho menos eficiente en presencia de una falta de coincidencia entre la plantilla y el cebador, por lo que el éxito de amplificación con un cebador señales SNP específico presencia del SNP específico en una secuencia. Ver SNP genotipo para más información.
- Asamblea PCR o la Polimerasa Asamblea Ciclismo (PCA): PCR Asamblea es la síntesis artificial de secuencias de ADN largas mediante la realización de PCR en un grupo de oligonucleótidos largos con segmentos superpuestos cortos. Los oligonucleótidos alternos entre las direcciones sentido y antisentido, y los segmentos superpuestos determinar el orden de los fragmentos de PCR de ese modo producir selectivamente el producto final de ADN de largo.
- PCR asimétrica: PCR asimétrica se utiliza para amplificar preferentemente una cadena de la original de ADN más que el otro. Se encuentra uso en algunos tipos de secuenciación e hibridación de sondeo donde habiendo sólo se requiere uno de los dos stands complementarios. PCR se lleva a cabo como de costumbre, pero con un gran exceso de los cebadores para la cadena elegido. Debido a la lenta ( aritmética ) posterior amplificación en la reacción después de que el cebador limitante se ha utilizado hasta, se requieren ciclos adicionales de PCR. Una modificación reciente en este proceso, conocido como L inear- A fter- T he- E xponential-PCR (LATE-PCR), utiliza un cebador limitativo con una temperatura más alta de fusión ( Temperatura de fusión | Tm) que el exceso de imprimación para mantener la eficiencia de la reacción como la concentración del cebador limitante disminuye mediados de reacción.
- Amplificación dependiente de helicasa: Esta técnica es similar a la PCR tradicional, pero utiliza una temperatura constante en lugar de bicicleta a través de ciclos de desnaturalización y reasociación / extensión. DNA helicasa, una enzima que se desenrolla de ADN, se utiliza en lugar de la desnaturalización térmica.
- PCR de arranque en caliente: Esta es una técnica que reduce la amplificación no específica durante la configuración inicial de las etapas de la PCR. La técnica se puede realizar manualmente mediante el calentamiento de los componentes de reacción a la temperatura de fusión (por ejemplo, 95ºC) antes de añadir la polimerasa. Sistemas enzimáticos especializados se han desarrollado que inhiben la actividad de la polimerasa a temperatura ambiente, ya sea por la unión de un anticuerpo o por la presencia de inhibidores unidos covalentemente que sólo se disocian después de una etapa de activación a alta temperatura. De arranque en caliente / frío con acabado PCR se consigue con nuevas polimerasas híbridas que son inactivos a temperatura ambiente y se activan de inmediato a la temperatura de alargamiento.
- Intersequence-específica (ISSR) PCR: un método de PCR para la toma de huellas dactilares de ADN que amplifica las regiones entre algunas secuencias simples repetidas para producir una huella digital única de longitudes de los fragmentos amplificados.
- PCR inverso: un método utilizado para permitir PCR cuando se conoce sólo una secuencia interna. Esto es especialmente útil en la identificación de secuencias que flanquean a varios insertos genómicos. Esto implica una serie de Digestiones de ADN y auto ligadura, resultando en secuencias conocidas en cualquiera de los extremos de la secuencia desconocida.
- La ligación mediada por PCR: Este método utiliza pequeñas enlazadores de ADN se ligó al ADN de interés y múltiples cebadores de recocido para los enlazadores de ADN; se ha utilizado para la secuenciación de ADN, el genoma caminar, y Footprinting ADN.
- PCR específica de metilación (MSP): El método MSP fue desarrollado por Stephen Baylin y Jim Herman en la Escuela de Medicina Johns Hopkins, y se utiliza para detectar la metilación de las islas CpG en el ADN genómico. ADN se trata primero con bisulfito de sodio, que convierte las bases de citosina no metiladas en uracilo, que es reconocido por los cebadores de PCR como timina. Dos PCR se llevan a cabo entonces en el ADN modificado, usando el cebador conjuntos idénticos excepto en las islas CpG dentro de las secuencias de cebadores. En estos puntos, uno conjunto de cebadores reconoce ADN con citosinas para amplificar el ADN metilado, y un conjunto reconoce ADN con uracilo o timina para amplificar el ADN no metilado. MSP mediante qPCR también se puede realizar para obtener información cuantitativa y no cualitativa sobre la metilación.
- Miniprimer PCR: Miniprimer PCR utiliza una novela polimerasa termoestable (S-TBR) que puede extenderse desde cebadores cortos ("smalligos") tan corto como 9 o 10 nucleótidos, en lugar de los aproximadamente 20 nucleótidos requeridos por Taq. Este método permite la orientación de PCR más pequeño regiones de unión al cebador, y es particularmente útil para amplificar desconocido, pero conserva, secuencias de ADN, tales como los 16S (o 18S rRNA genes eucariotas). 16S rRNA miniprimer PCR se utilizó para caracterizar una comunidad tapete microbiano que crece en un ambiente extremo, un estanque hipersalinos en Puerto Rico. En ese estudio, profundamente divergentes secuencias fueron descubiertos con alta frecuencia e incluyeron representantes que de fi ne dos nuevos taxones a nivel de división, lo que sugiere que la PCR miniprimer puede revelar nuevas dimensiones de la diversidad microbiana. Al ampliar el "espacio de secuencia" que puede ser consultada por los cebadores de PCR, esta técnica puede permitir estrategias de PCR novedosos que no son posibles dentro de los límites del primer diseño impuestas por Taq y otras enzimas de uso común.
- Multiplex Ligadura dependiente de la sonda de amplificación (MLPA): permite múltiples objetivos a amplificar con un único par de cebadores, evitando así las limitaciones de resolución de PCR multiplex (véase más adelante).
- Multiplex-PCR: El uso de múltiples conjuntos de cebadores únicos dentro de una única mezcla de PCR para producir amplicones de diferentes tamaños específicos para diferentes secuencias de ADN. Al dirigirse a múltiples genes a la vez, la información adicional puede ser obtenida a partir de una única prueba que de otra manera requeriría varias veces los reactivos y más tiempo para llevar a cabo. Temperaturas de recocido para cada uno de los conjuntos de cebadores deben ser optimizadas para funcionar correctamente dentro de una sola reacción, y tamaños de amplificación, es decir, su longitud de pares de bases, deben ser lo suficientemente diferente para formar bandas distintas cuando se visualizan por electroforesis en gel.
- Nested PCR: aumenta la especificidad de la amplificación de ADN, mediante la reducción de fondo debido a la amplificación no específica de ADN. Dos conjuntos de cebadores se utilizan en dos PCR sucesivas. En la primera reacción, un par de cebadores se usa para generar productos de ADN, que además del objetivo previsto, todavía pueden consistir en fragmentos de ADN amplificados no específicamente. El producto (s) se utiliza a continuación en una segunda PCR con un conjunto de cebadores cuyos sitios de unión son completamente o parcialmente diferente de y situado 3 'de cada uno de los cebadores utilizados en la primera reacción. Nested PCR es a menudo más éxito en la amplificación de fragmentos de ADN específicamente largas que la PCR convencional, pero requiere un conocimiento más detallado de las secuencias diana.
- Superposición PCR de extensión: es un técnica de ingeniería genética que permite la construcción de una secuencia de ADN con una alteración insertado más allá del límite de la más larga longitud del cebador práctico.
- PCR cuantitativa (Q-PCR): se utiliza para medir la cantidad de un producto de PCR (preferiblemente en tiempo real). Es el método de elección para medir cuantitativamente las cantidades de partida de ADN, ADNc o ARN. Q-PCR se utiliza comúnmente para determinar si una secuencia de ADN está presente en una muestra y el número de sus copias en la muestra. El método actualmente con el más alto nivel de precisión es la PCR cuantitativa en tiempo real. A menudo se conoce como confusamente RT-PCR (R eal T ime PCR) o RQ-PCR. QRT-PCR o RTQ-PCR son contracciones más apropiadas. RT-PCR se refiere comúnmente a transcripción inversa PCR (véase más adelante), que se utiliza a menudo en conjunción con Q-PCR. Métodos de QRT-PCR usan colorantes fluorescentes, tales como SYBR Green, o Las sondas de ADN que contiene fluoróforo, tales como TaqMan, para medir la cantidad de producto amplificado en tiempo real.
- RT-PCR: (R everse T ranscription PCR) es un método utilizado para amplificar, aislar o identificar una secuencia conocida de un celular o tejido RNA. La PCR es precedida por una reacción utilizando la transcriptasa inversa para convertir ARN para cDNA. RT-PCR es ampliamente utilizado en de perfiles de expresión, para determinar la expresión de un gen o para identificar la secuencia de un transcrito de ARN, incluyendo inicio de la transcripción y sitios de terminación y, si se conoce la secuencia de ADN genómico de un gen, para mapear la ubicación de exones y intrones en el gen. El extremo 5 'de un gen (que corresponde al sitio de inicio de transcripción) se identifican típicamente por un método de RT-PCR, nombrado RACE-PCR, la abreviatura de rápida amplificación de cDNA Ends.
- Solid PCR Fase: abarca múltiples significados, incluyendo Polony amplificación (donde las colonias de PCR se derivan en una matriz de gel, por ejemplo), 'Puente de PCR' (los únicos cebadores presente están unidos covalentemente a la superficie de soporte sólido), en fase sólida convencional PCR (donde se aplica PCR asimétrica en presencia de un sólido apoyo imprimación rodamiento con secuencia de búsqueda de uno de los cebadores acuosas) y mejorada en fase sólida PCR (donde PCR convencional en fase sólida se puede mejorar mediante el empleo de alta imprimación soporte sólido Tm con la aplicación de un "paso 'térmica para favorecer soporte sólido de cebado).
- TAIL-PCR: PCR asimétrica entrelazado térmica se utiliza para aislar secuencia desconocida que flanquean una secuencia conocida. Dentro de la secuencia conocida TAIL-PCR utiliza un par de cebadores anidados con diferentes temperaturas de recocido; un cebador degenerado se utiliza para amplificar en la otra dirección de la secuencia desconocida.
- Touchdown PCR: una variante de la PCR que tiene como objetivo reducir el fondo no específica mediante la reducción gradual de la temperatura de recocido como ciclismo PCR progresa. La temperatura de hibridación en los ciclos iniciales suele ser de unos pocos grados (C3-5) por encima de la Tm de los cebadores utilizados, mientras que en los ciclos posteriores, que está a pocos grados (C3-5) por debajo de la imprimación T m. Las temperaturas más altas dan una mayor especificidad para la unión del cebador, y las temperaturas más bajas permiten la amplificación más eficiente de los productos específicos formados durante los ciclos iniciales.
- PAN-AC: Este método utiliza condiciones isotérmicas para la amplificación, y puede ser utilizado en las células vivas.
- Universal caminar rápido: este método permite genoma caminar y la huella genética utilizando una más específica 'de dos caras' PCR que los enfoques convencionales "de una cara" (usando sólo un cebador específico de gen y un cebador en general - que puede conducir a "ruido artefactual ') en virtud de un mecanismo que implica la formación de la estructura de lazo. Derivados simplificados de UFW son RAGE Lane (termina lazo dependiente PCR anidada para la rápida amplificación de ADN genómico), 5'RACE Lane y 3'RACE Lane.
Historia
Un documento de 1971 en el Journal of Molecular Biology por Kleppe y colaboradores describe un método que utiliza primero un ensayo enzimático para replicar una plantilla corta de ADN con cebadores in vitro. Sin embargo, esta manifestación temprana del principio básico de PCR no recibió mucha atención, y la invención de la reacción en cadena de la polimerasa en 1983 se acredita generalmente a Kary Mullis.
En el núcleo del método de PCR es el uso de un adecuado ADN polimerasa capaz de soportar las altas temperaturas de> 90 ° C (> 195 ° F) necesarios para la separación de las dos hebras de ADN en el ADN de doble hélice después de cada ciclo de replicación. Las ADN polimerasas empleadas inicialmente para experimentos in vitro presagian PCR fueron incapaces de resistir estas altas temperaturas. Así que los primeros procedimientos para la replicación del ADN eran muy ineficiente, consume tiempo, y requieren grandes cantidades de ADN polimerasa y la manipulación continua durante todo el proceso.
Un descubrimiento de 1976 Polimerasa Taq una polimerasa de ADN purificado a partir de la bacteria termófila, Thermus aquaticus, que se produce naturalmente en caliente (50 a 80 ° C (120-175 ° F)) entornos allanó el camino para mejoras dramáticas del método de PCR. La polimerasa de ADN aislado de T. aquaticus es estable a altas temperaturas que permanecen activas incluso después de la desnaturalización del ADN, obviando así la necesidad de añadir nuevo ADN polimerasa después de cada ciclo. Esto permitió un proceso basado en el termociclador automatizado para la amplificación de ADN.
En el momento que desarrolló la PCR en 1983, Mullis trabajaba en Emeryville, California Cetus Corporation, una de las primeras biotecnológicas empresas. Allí, él fue el responsable de la síntesis de cadenas cortas de ADN. Mullis ha escrito que él concibió de PCR durante la navegación a lo largo del Pacific Coast Highway una noche en su coche. Estaba jugando en su mente con una nueva forma de analizar los cambios (mutaciones) en el ADN cuando se dio cuenta de que en lugar de eso había inventado un método para amplificar cualquier región de ADN a través de ciclos repetidos de duplicación impulsado por la ADN polimerasa.
En Scientific American, Mullis resumió el procedimiento: "A partir de una sola molécula de ADN material genético, la PCR puede generar 100 mil millones de moléculas similares en una tarde, la reacción es fácil de ejecutar No requiere más que un tubo de ensayo, entre otros.. reactivos simples, y una fuente de calor ". Fue galardonado con el Premio Nobel de Química en 1993 por su invención, siete años después de que él y sus colegas del Cetus primero puso su propuesta a la práctica. Sin embargo, algunas controversias se han mantenido sobre las contribuciones intelectuales y prácticas de otros científicos al trabajo Mullis ', y si él había sido el único inventor del principio de PCR. (Ver artículo principal: Kary Mullis)
Guerras de patentes
La técnica de PCR fue patentado por Cetus Corporation, donde Mullis trabajaba cuando inventó la técnica en 1983. La enzima Taq polimerasa también fue cubierto por patentes. Ha habido varios juicios de alto perfil relacionados con la técnica, incluyendo un pleito fracasado interpuesto por DuPont. La compañía farmacéutica Hoffmann-La Roche adquirió los derechos sobre las patentes en 1992 y actualmente tiene los que todavía están protegidos.
Una batalla de patentes relacionadas con el enzima Taq polimerasa está todavía en curso en varias jurisdicciones de todo el mundo entre Roche y Promega. Los argumentos jurídicos se han extendido más allá de la vida de las patentes originales polimerasa PCR y Taq, que expiró el 28 de marzo de 2005