

L'équilibre chimique
Contexte des écoles Wikipédia
Cette sélection de wikipedia a été choisi par des bénévoles aidant les enfants SOS de Wikipedia pour cette sélection Wikipedia pour les écoles. SOS Children travaille dans 45 pays africains; pouvez-vous aider un enfant en Afrique ?
Dans un processus chimique, équilibre chimique est l'état dans lequel le activités chimiques ou les concentrations des réactifs et des produits ne ont aucun changement net dans le temps. Habituellement, ce serait l'état qui résulte de l'avant processus chimique procède au même rythme que leur réaction inverse. Les taux de réaction de la marche avant et arrière réactions sont généralement pas à zéro, mais, étant égales par ailleurs, il n'y a aucun changement net dans l'un des réactifs ou de produits concentrations. Ce processus est appelé équilibre dynamique
| |||||
Introduction

Dans une réaction chimique , lorsque les réactifs sont mélangés ensemble dans un récipient de réaction (et chauffée si nécessaire), l'ensemble des corps réactionnels ne est pas converti en les produits. Après un certain temps (qui peut être plus courte que millionièmes de seconde ou plus que l'âge de l'univers), il viendra un moment où un montant fixe de réactifs existera en harmonie avec une quantité fixe de produits, les quantités de changement ni plus. Ce est ce qu'on appelle l'équilibre chimique.
Le concept de l'équilibre chimique a été développé après Berthollet (1803) a constaté que certaines réactions chimiques sont réversible. Pour toute réaction, tel que

être à l'équilibre les taux des réactions avant et arrière (arrière) doivent être égaux. Dans ce équation chimique avec des flèches pointant de harpons dans les deux sens pour indiquer l'équilibre, A et B sont réactif espèce chimique, S et T sont des espèces de produits, et α, β, σ, et τ sont les les coefficients stoechiométriques des réactifs et des produits respectifs. La position d'une réaction d'équilibre est dit de mentir loin vers la droite si, à l'équilibre, presque tous les réactifs sont utilisés et loin vers la gauche si guère produit est formé à partir des réactifs.
Guldberg et Waage (1865), se appuyant sur les idées de Berthollet, a proposé la la loi d'action de masse:


où A, B, S et T sont masses actives et k + et k - sont les constantes de vitesse. Depuis l'avant et vers l'arrière taux sont égaux:
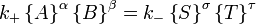
et le rapport des constantes de vitesse est également constante, maintenant connu sous le nom la constante d'équilibre.
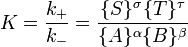
Par convention les produits constituent le numérateur . Cependant, la la loi d'action de masse ne est valable que pour les réactions en une étape concertées qui procèdent par un seul transition Etat et ne est pas valable en général, car les équations de taux ne, en général, suivent pas la stoechiométrie de la réaction comme Guldberg et Waage a proposé (voir, par exemple, la substitution nucléophile aliphatique par réaction SN1 ou de l'hydrogène et du brome pour former le bromure d'hydrogène). Egalité des taux à terme et de réaction en amont, cependant, est un condition nécessaire à l'équilibre chimique, mais il ne est pas suffisante pour expliquer pourquoi l'équilibre se produit.
Malgré l'échec de cette dérivation, la constante d'équilibre pour la réaction est en effet une constante, indépendante des activités des différentes espèces concernées, si elle ne dépend que de la température observée par le Relation de Van't Hoff. L'ajout d'un catalyseur aura une incidence sur la réaction à la fois vers l'avant et la réaction inverse de la même façon et ne aura pas d'effet sur la constante d'équilibre. Le catalyseur va accélérer deux réactions augmentant ainsi la vitesse à laquelle l'équilibre est atteint.
Bien que les concentrations à l'équilibre macroscopiques sont constantes dans le temps des réactions se produisent au niveau moléculaire. Par exemple, dans le cas de l'acide éthanoïque dissous dans l'eau et formant éthanoate et ions hydronium,
- CH 3 CO 2 H + H 2 O ⇌ CH 3 CO 2 - + H 3 O +
un proton peut sauter d'une molécule d'acide éthanoïque à une molécule d'eau, puis à un ion éthanoate pour former une autre molécule d'acide éthanoïque et en laissant le nombre de molécules d'Acide acétique inchangé. Ceci est un exemple de équilibre dynamique. Équilibres, comme le reste de la thermodynamique, sont des phénomènes statistiques, moyennes de comportement microscopique.
Le principe de Le Chatelier (1884) est un principe utile qui donne une idée qualitative de la réponse d'un système à l'équilibre à des changements dans les conditions de réaction. Si un équilibre dynamique est perturbée par le changement des conditions, la position d'équilibre se déplace pour contrer le changement. Par exemple, en ajoutant plus S de l'extérieur feront apparaître un excédent de produits, et le système va essayer de contrer ce en augmentant la réaction inverse et en poussant le point d'équilibre vers l'arrière (si la constante d'équilibre restera le même).
Si l'acide minéral est ajouté au mélange de l'acide éthanoïque, l'augmentation de la concentration de l'ion hydronium, la quantité de dissociation doit diminuer à mesure que la réaction est entraînée vers la gauche conformément à ce principe. Cela peut aussi être déduite de l'expression de la constante d'équilibre pour la réaction:

si {H 3 O +} {augmente CH 3 CO 2 H} {doit augmenter et CH 3 CO 2 -} doit diminuer.
Une version quantitative est donnée par la quotient de réaction.
JW Gibbs a suggéré en 1873 que l'équilibre est atteint lorsque le Enthalpie libre du système est à sa valeur minimale (en supposant que la réaction est effectuée sous une pression constante). Ce qui veut dire que la dérivée de l'énergie de Gibbs par rapport à réaction de coordonnées (une mesure de la ampleur de la réaction qui a eu lieu, allant de zéro pour tous les réactifs à un maximum pour tous les produits) disparaît, signalant un point fixe. Ce dérivé est généralement appelé, pour certaines raisons techniques, le changement d'énergie Gibbs. Ce critère est à la fois nécessaire et suffisante. Si un mélange ne est pas à l'équilibre, la libération de l'énergie d'excès de Gibbs (ou Helmholtz énergie à des réactions à volume constant) est la «force motrice» pour la composition du mélange de changer jusqu'à ce qu'un équilibre soit atteint. La constante d'équilibre peut être liée à la norme énergie de Gibbs changement pour la réaction par l'équation

où R est le constante universelle des gaz et T la température .
Lorsque les réactifs sont dissous dans un milieu de haut la force ionique du quotient de les coefficients d'activité peuvent être prises pour être constante. Dans ce cas, le quotient de la concentration, K c,
![K_C = \ frac {[S] ^ \ sigma [T] ^ \ tau} {[A] ^ \ alpha [B] ^ \ beta}](../../images/117/11709.png)
où [A] est la concentration de A, etc., est indépendante de la concentration des réactifs d'analyse. Pour cette raison, les constantes d'équilibre pour les solutions sont habituellement déterminé dans les médias de grande force ionique. C K varie en fonction de la force ionique, la température et la pression (ou le volume). De même, K p pour les gaz dépend de pression partielle. Ces constantes sont plus faciles à mesurer et rencontrés dans les cours de chimie du secondaire.
Thermodynamique
La relation entre l'énergie de Gibbs, et la constante d'équilibre peut être trouvée en prenant en compte les potentiels chimiques. La thermodynamique condition d'équilibre chimique est
- À pression constante AG = 0 (AG est le changement énergie libre de Gibbs pour la réaction)
- A Aa à volume constant = 0 (Aa est le changement de Énergie libre pour la réaction)
Dans cet article, seul le cas de pression constante est considérée. Le cas de volume constant est important dans géochimie et chimie de l'atmosphère, où les variations de pression sont importants. Notez que, si des réactifs et des produits étaient en état standard (complètement pure), alors il n'y aurait pas de réversibilité et pas d'équilibre. Le mélange des produits et réactifs contribue une grande entropie (connu sous le nom entropie de mélange) aux Etats contenant mélange égal de produits et réactifs. La combinaison du changement standard énergie de Gibbs et l'énergie de Gibbs de mélange détermine l'état d'équilibre.
En général, un système d'équilibre est définie par l'écriture d'une équation d'équilibre de la réaction
Afin de satisfaire à la condition thermodynamique pour l'équilibre, l'énergie de Gibbs doit être immobile, ce qui signifie que la dérivée de G par rapport à coordonnée de réaction (AG) doit être zéro. On peut montrer que Ag est, en fait, égale à la différence entre la les potentiels chimiques des produits et ceux des réactifs. Par conséquent, la somme des énergies de Gibbs des réactifs doit être égale à la somme des énergies de Gibbs des produits.
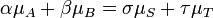
où μ est dans ce cas une molaire partielle d'énergie Gibbs, un le potentiel chimique. Le potentiel chimique d'un réactif A est une fonction de la activité, {A} de ce réactif.
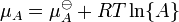
En substituant les expressions de ce genre dans l' équation de l'énergie Gibbs :
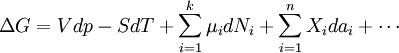
, à la pression et la température constante devient:
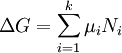
résulte en:
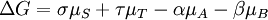
En substituant les potentiels chimiques:
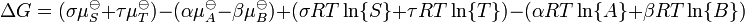
la relation devient:
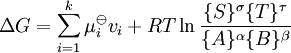
A l'équilibre, 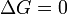 et donc
et donc
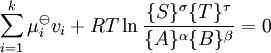
menant à:
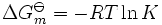
Ag O m est la molaire Gibbs changement d'énergie standard pour la réaction et K est le la constante d'équilibre. A noter que les activités et les constantes d'équilibre sont des nombres sans dimension.
Traitement de l'activité
L'expression de la constante d'équilibre peut être réécrite comme étant le produit d'un quotient de la concentration, K c et un activité coefficient quotient, Γ.
![K = \ frac {{[S]} ^ \ sigma {[T]} ^ \ tau ...} {{[A]} ^ \ alpha {[B]} ^ \ beta ...} \ times \ frac {{\ gamma_S} ^ \ sigma {\ gamma_T} ^ \ tau ...} {{\ gamma_A} ^ \ alpha {\ gamma_B} ^ \ beta ...} = K_C \ Gamma](../../images/117/11719.png)
[A] est la concentration du réactif A, etc. Il est possible en principe d'obtenir des valeurs de coefficients d'activité, γ. Pour les solutions, les équations comme la Force ionique ou extensions comme Davies équation ou les équations Pitzer peuvent être utilisés. Logiciel (ci-dessous). Cependant ce ne est pas toujours possible. Il est courant de penser que Γ est une constante, et d'utiliser le quotient de la concentration en place de la constante d'équilibre thermodynamique. Il est également pratique générale à utiliser le terme constant de l'équilibre au lieu de la concentration quotient plus précis. Cette pratique sera suivie ici.
Pour les réactions en phase gazeuse, pression partielle est utilisée à la place de la concentration et coefficient de fugacité en place du coefficient d'activité. Dans le monde réel, par exemple, lors de la prise l'ammoniac dans l'industrie, les coefficients de fugacité doit être prise en compte. Fugacité, f, est le produit de la pression partielle et le coefficient de fugacité. Le potentiel chimique d'une espèce dans la phase gazeuse est donnée par

si l'expression générale définissant une constante d'équilibre est valable pour les deux phases de la solution et de gaz.
Justification de l'utilisation des quotients de concentration
En solution aqueuse, les constantes d'équilibre sont habituellement déterminées en présence d'un électrolyte "inerte" tel que nitrate de sodium NaNO 3 ou Le perchlorate de potassium KClO 4. Le la force ionique, I, d'une solution contenant un sel dissous, X + Y -, est donnée par
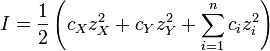
où c représente la concentration, z signifie charge ionique et la somme est prise sur toutes les espèces en équilibre. Lorsque la concentration du sel dissous est beaucoup plus élevée que les concentrations des réactifs d'analyse, la force ionique est effectivement constante. Étant donné que les coefficients d'activité dépendent de la force ionique des coefficients d'activité de l'espèce sont effectivement indépendante de la concentration. Ainsi, l'hypothèse selon laquelle Γ est constante est justifiée. Le quotient de la concentration est un multiple simple de la constante d'équilibre.
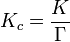
Toutefois, K c varie avec la force ionique. Si elle est mesurée à une série de forces ioniques différentes, la valeur peut être extrapolée à zéro de la force ionique. Le quotient de concentration obtenu de cette manière est connue, paradoxalement, à la constante d'équilibre thermodynamique.
Pour utiliser la valeur publiée d'une constante d'équilibre dans des conditions de force ionique différentes des conditions utilisées dans sa détermination, la valeur doit être ajustée Logiciel (ci-dessous).
Mélanges métastables
Un mélange peut être ne semblent pas avoir tendance à changer, mais il ne est pas à l'équilibre. Par exemple, un mélange de SO 2 et O 2 est métastable car il ya une barrière cinétique de formation du produit, SO 3.
- 2SO 2 + O 2
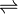 2SO 3
2SO 3
La barrière peut être surmontée quand un catalyseur est également présent dans le mélange comme dans le processus contacter, mais le catalyseur ne affecte pas les concentrations à l'équilibre.
De même, la formation de bicarbonate de dioxyde de carbone et eau est très lente dans des conditions normales
- CO 2 + 2H 2 O HCO 3 - + H 3 O +
mais presque instantanée en présence du catalyseur enzyme anhydrase carbonique.
Composés purs dans équilibres
Lorsque des substances pures (liquides ou solides) sont impliqués dans les équilibres qu'ils ne apparaissent pas dans l'équation d'équilibre
L'application de la formule générale pour une constante d'équilibre pour le cas spécifique de l'acide éthanoïque on obtient

![K_C = \ frac {[{} ^ CH_3CO_2 -] [{} ^ H_3O +]} {[{} CH_3CO_2H] [{} H_2O]}](../../images/117/11725.png)
On peut supposer que la concentration de l'eau est constante. Cette hypothèse sera solutions valables pour tous, mais très concentrés. L'expression constante d'équilibre est donc généralement écrit comme
![K = \ frac {[{} ^ CH_3CO_2 -] [{} ^ H_3O +]} {[{} CH_3CO_2H]}](../../images/117/11726.png)
où maintenant
![K = K_C * [H_2O] \,](../../images/117/11727.png)
un facteur constant est incorporé dans la constante d'équilibre.
Un cas particulier est le auto-ionisation d'elle-même de l'eau

La constante Autoprotolyse est défini comme
![K_w = [^ H +] [^ OH -] \,](../../images/117/11729.png)
Il est parfaitement légitime d'écrire [H +] pour le concentration d'ions hydronium, depuis l'état de la solvatation du proton est constante (en solution diluée) et ainsi ne affecte pas les concentrations à l'équilibre. K w varie avec la variation de la résistance et / ou la température ionique.
Les concentrations de H + et OH - ne sont pas des quantités indépendantes. Le plus souvent [OH -] est remplacé par K w [H +] -1 dans des expressions constantes d'équilibre qui, autrement, l'hydroxyde .
Solides également ne apparaissent pas dans l'équation d'équilibre. Un exemple est le Boudouard réaction:
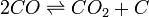
pour laquelle l'équation (sans carbone solide) se écrit:
![K_C = \ frac {[CO_2]} {[CO] ^ 2}](../../images/117/11731.png)
Équilibres multiples
Prenons le cas d'un acide dibasique H 2 A. Lorsqu'il est dissous dans l'eau, le mélange va contenir H 2 A, HA - et A 2. Cet équilibre peut être divisée en deux étapes, dans chacune desquelles un proton est libéré.
![H_2A \ rightleftharpoons HA ^ - ^ + H +: K_1 = \ frac {[HA ^ -] [^ H +]} {[H_2A]}](../../images/117/11732.png)
![HA ^ - \ rightleftharpoons A ^ {2-} ^ + H +: K_2 = \ frac {[A ^ {2 -}] [^ H +]} {[HA ^ -]}](../../images/117/11733.png)
K 1 et K 2 sont des exemples de constantes d'équilibre par étapes. La constante d'équilibre global, 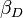 , Est produit par étapes des constantes.
, Est produit par étapes des constantes.
![A d'H_2A \ ^ {2-} + 2H + ^: \ beta_D = \ frac {[A ^ {2 -}] [^ H +] ^ 2} {[H_2A]} = K_1K_2](../../images/117/11735.png)
On notera que ces constantes sont constantes de dissociation parce que les produits sur le côté droit de l'expression d'équilibre sont des produits de dissociation. Dans de nombreux systèmes, il est préférable d'utiliser des constantes d'association.
![A ^ {2-} + H ^ + \ rightleftharpoons HA ^ -: \ beta_1 = \ frac {[HA ^ -]} {[A ^ {2 -}] [^ H +]}](../../images/117/11736.png)
![A ^ {2-} + 2H ^ + \ rightleftharpoons H_2A: \ beta_2 = \ frac {[H_2A]} {[A ^ {2 -}] [^ H +] ^ 2}](../../images/117/11737.png)
β 1 et β 2 sont des exemples de constantes d'association. Il est clair que β 1 = 1 / K 2 et β 2 = 1 / β D; lg = pK β 1 et 2 β 2 = lg pK 2 + pK 1
Effet de la variation de température sur une constante d'équilibre
L'effet des changements de température sur une constante d'équilibre est donné par la Relation de Van't Hoff
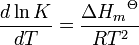
Ainsi, par réactions exothermiques, (AH est négatif) K diminue avec la température, mais, pour réactions endothermiques, (AH est positif) K augmente avec la température. Une formulation alternative est
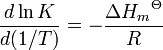
A première vue, cela semble offrir un moyen d'obtention de la norme enthalpie molaire de la réaction par l'étude de la variation de K avec la température. En pratique, cependant, le procédé ne est pas fiable parce que la propagation d'erreur donne presque toujours de très grandes erreurs sur les valeurs calculées de cette façon.
Types d'équilibre et certaines applications
- Dans la phase gazeuse. Les moteurs de fusée
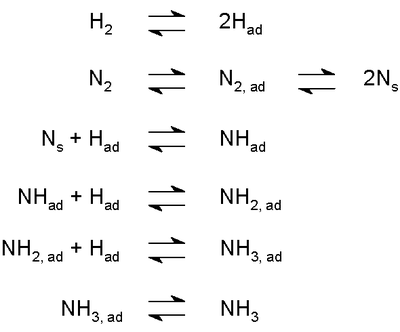
- La synthèse industrielle tels que l'ammoniac dans le Procédé Haber-Bosch (représenté à droite) a lieu à travers une succession d'étapes d'équilibre, y compris les processus de l'adsorption.
- la chimie atmosphérique
- L'eau de mer et d'autres eaux naturelles: Océanographie chimique
- La distribution entre deux phases
- Coefficient logD-Distribution: Important pour les produits pharmaceutiques où lipophile est une propriété importante d'un médicament
- L'extraction liquide-liquide, L'échange d'ions, la chromatographie
- produit de solubilité
- Absorption et la libération de l'oxygène par hémoglobine dans le sang
- Équilibres acide / base: Acid constante de dissociation, hydrolyse, des solutions tampons, indicateurs, homéostasie acido-basique
- Métal-ligand de complexation: des agents séquestrants, la thérapie de chélation, Réactifs de contraste IRM, Schlenk équilibre
- La formation d'adduits: La chimie hôte-invité, la chimie supramoléculaire, reconnaissance moléculaire, tétroxyde de diazote
- Dans certaines réactions oscillants, l'approche à l'équilibre ne est pas asymptotiquement mais sous la forme d'une oscillation amortie.
- La connexes Équation de Nernst en électrochimie donne la différence de potentiel d'électrode en fonction de la concentration d'oxydo-réduction.
- Lorsque les molécules de chaque côté de l'équilibre sont capables de réagir de manière irréversible en outre dans des réactions secondaires, le rapport final du produit est déterminé en fonction de la Curtin-Hammett principe.
Dans ces applications, des termes tels que la stabilité constante, constante de formation, constante de liaison, constante d'affinité, d'association / constante de dissociation sont utilisés. En biochimie, il est courant de donner unités pour les constantes de liaison, qui servent à définir les unités de concentration utilisées lorsque la valeur de la constante a été déterminée.
Composition d'un mélange à l'équilibre
Quand le seul équilibre est celui de la formation d'un adduit 1: 1 comme la composition d'un mélange, il existe un certain nombre de façons dont la composition d'un mélange peut être calculée. Par exemple, voir table de glace pour une méthode traditionnelle de calcul du pH d'une solution d'un acide faible.
Il existe trois approches pour le calcul générale de la composition d'un mélange à l'équilibre.
- L'approche la plus fondamentale est de manipuler les différentes constantes d'équilibre jusqu'à ce que les concentrations désirées sont exprimés en termes de constantes d'équilibre mesurées (équivalent à la mesure de potentiels chimiques) et les conditions initiales.
- Minimiser l'énergie Gibbs du système.
- Satisfaire l'équation de bilan de masse. Les équations de bilan de masse sont tout simplement des déclarations qui démontrent que la concentration totale de chaque réactif doit être constante par la loi du conservation de la masse.
Résolvant les équations de bilan massique
En général, les calculs sont un peu compliqué. Par exemple, dans le cas d'un acide dibasique, H 2 A dissous dans l'eau les deux corps réactionnels peuvent être spécifiés comme le de base conjugué, A 2, et de la protons, H +. Les équations suivantes de bilan de masse pourraient se appliquer tout aussi bien à une base telle que Le 1,2-diaminoéthane, auquel cas la base elle-même est désigné comme réactif A:
![T_A = [A] + [HA] + [H_2A] \,](../../images/117/11744.png)
![T_H = [H] + [HA] + 2 [H_2A] - [OH] \,](../../images/117/11745.png)
Avec T A la concentration totale des espèces A. A noter qu'il est d'usage d'omettre les charges ioniques lors de l'écriture et l'utilisation de ces équations.
Lorsque les constantes d'équilibre sont connus et les concentrations totales sont spécifiés il ya deux équations à deux "concentrations libres" inconnus [A] et [H]. Cela découle du fait que [HA] = β 1 [A] [H], [H 2 A] = β 2 [A] [H] 2 et [OH] = K w [H] -1
![T_A = [A] + \ beta_1 [A] [H] + \ beta_2 [A] [H] ^ 2 \,](../../images/117/11746.png)
![T_H = [H] + \ beta_1 [A] [H] + 2 \ beta_2 [A] [H] ^ 2 - K_w [H] ^ {- 1} \,](../../images/117/11747.png)
de sorte que les concentrations des "complexes" sont calculés à partir des concentrations libres et les constantes d'équilibre. Expressions générales applicables à tous les systèmes avec deux réactifs, A et B seraient
![T_A = [A] + \ {sum_i p_i \ beta_i [A] ^ {} p_i [B] ^ {}} Q_i](../../images/117/11748.png)
![T_B = [B] + \ {sum_i Q_i \ beta_i [A] ^ {} p_i [B] ^ {}} Q_i](../../images/117/11749.png)
Il est facile de voir comment cela peut être portée à trois ou plusieurs réactifs.
Composition de polyacides en tant que fonction du pH
Composition selon l'une des solutions contenant des réactifs A et H est facile de calculer en fonction de p [H]. Lorsque [H] est connu, la concentration libre [A] est calculée à partir de l'équation du bilan massique dans A. Voici un exemple des résultats qui peuvent être obtenus.

Ce schéma, pour l'hydrolyse de l' aluminium Acide de Lewis Al 3+ aq montre les concentrations d'espèces pour une solution 5 M × 10 -6 d'un sel d'aluminium en tant que fonction du pH. Chaque concentration est indiquée en pourcentage de l'aluminium total.
équilibres de solution avec précipitations
Le diagramme ci-dessus illustre le point que précipiter ce ne est pas l'une des principales espèces dans l'équilibre de la solution peut être formée. A pH 5,5 juste au-dessous les principales espèces présentes dans une solution 5 uM de Al 3+ sont des hydroxydes d'aluminium Al (OH) 2+, Al (OH) 2 + et Al 13 (OH) 32 7+, mais sur l'élévation du pH Al (OH) 3 précipité de la solution. Cela se produit parce que Al (OH) 3 a une très grande énergie treillis. Lorsque le pH se élève de plus en plus Al (OH) 3 sort de la solution. Ceci est un exemple de Le principe de Le Chatelier en action: L'augmentation de la concentration de l'ion hydroxyde provoque plus de l'hydroxyde d'aluminium pour précipiter, ce qui élimine l'hydroxyde de la solution. Lorsque la concentration en soude est suffisamment élevée l'aluminate soluble, Al (OH) 4 -, est formée.
Un autre exemple commun où se produit la précipitation est quand un cation métallique interagit avec un ligand anionique pour former un complexe électriquement neutre. Si le complexe est hydrophopbic, il va précipiter hors de l'eau. Cela se produit avec le nickel ions Ni 2+ et diméthylglyoxime, (dmgH 2): dans ce cas l'énergie de réseau du solide ne est pas particulièrement grande, mais il dépasse largement l'énergie la solvatation de la molécule Ni (dmgH) 2.
Minimisation de l'énergie Gibbs
A l'équilibre, G est au minimum:

Pour un système fermé, pas de particules peuvent entrer ou sortir, même se ils peuvent se combiner de différentes façons. Le nombre total d'atomes de chaque élément restera constant. Cela signifie que la réduction ci-dessus doivent être soumis à des contraintes:
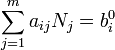
où  est le nombre d'atomes de l'élément i dans la molécule et b j i 0 est le nombre total d'atomes de l'élément i, qui est une constante, étant donné que le système est fermé. Se il ya un total de types d'atomes dans le système k, alors il y aura k ces équations.
est le nombre d'atomes de l'élément i dans la molécule et b j i 0 est le nombre total d'atomes de l'élément i, qui est une constante, étant donné que le système est fermé. Se il ya un total de types d'atomes dans le système k, alors il y aura k ces équations.
Ce est un problème classique dans optimisation, appelée minimisation sous contrainte. La méthode la plus courante de la résolution qu'il utilise le procédé de Multiplicateurs de Lagrange, aussi connu comme multiplicateurs indéterminé (bien que d'autres procédés peuvent être utilisés).
Définir:
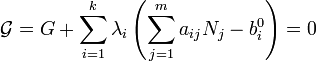
où le 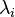 sont des multiplicateurs de Lagrange, une pour chaque élément. Cela permet à chacun de la
sont des multiplicateurs de Lagrange, une pour chaque élément. Cela permet à chacun de la  à traiter de façon indépendante, et il peut être démontré en utilisant les outils de calcul à variables multiples que la condition d'équilibre est donnée par
à traiter de façon indépendante, et il peut être démontré en utilisant les outils de calcul à variables multiples que la condition d'équilibre est donnée par
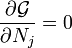 et
et 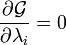
(Pour preuve voir Multiplicateurs de Lagrange)
Il se agit d'un ensemble d'équations inconnues à (m + k) (m + k) (la et le ) Et peut donc être résolu pour les concentrations à l'équilibre pour autant que les potentiels chimiques sont connus en tant que fonctions des concentrations à la température et une pression données. (Voir Bases de données thermodynamiques pour les substances pures).
Cette méthode de calcul des concentrations chimiques d'équilibre est utile pour les systèmes avec un grand nombre de molécules différentes. L'utilisation des équations de conservation atomiques de l'élément k de la contrainte de masse est simple, et remplace l'utilisation des équations des coefficients stoechiométriques.