

Énergie libre de Gibbs
À propos de ce écoles sélection Wikipedia
SOS Enfants produite ce site pour les écoles ainsi que ce site de vidéo sur l'Afrique . SOS Enfants a regardé des enfants en Afrique depuis quarante ans. Pouvez-vous aider dans leur travail en Afrique ?
| Mécanique statistique |
|---|
 |
|
Statistiques de particules
|
Ensembles
|
|
Modèles
|
Potentiels
|
Dans la thermodynamique , l'énergie libre de Gibbs ( Dénomination de l'UICPA recommandée: Gibbs l'énergie ou la fonction de Gibbs) est un potentiel thermodynamique qui mesure le travail «utile» ou un processus initiant obtenue à partir d'un isotherme, isobare système thermodynamique. Techniquement, l'énergie libre de Gibbs est la quantité maximale de travail non-expansion qui peut être extrait d'un système fermé ou ce maximum peut être atteint que dans un tout processus réversible. Lorsqu'un système passe d'un état initial bien défini à un état final bien défini, le Gibbs Ag de l'énergie libre est égal au travail échangée par le système avec son environnement, moins le travail des forces de pression, lors d'une transformation réversible du système du même état initial dans le même état final.
Énergie de Gibbs est également le potentiel chimique qui est minimisée quand un système atteint l'équilibre à la pression et la température constante. En tant que tel, ce est un critère pratique de la spontanéité de processus à pression et température constante.
L'énergie libre de Gibbs, appelé à l'origine de l'énergie disponible, a été développé dans les années 1870 par le physicien et mathématicien américain Willard Gibbs . En 1873, dans une note, Gibbs défini ce qu'il a appelé «l'énergie disponible" d'un corps en tant que telle:
| " | La plus grande quantité de travail mécanique qui peut être obtenu à partir d'une quantité donnée d'une certaine substance dans un état initial donné, sans augmenter son total de volumes ou de permettre à la chaleur de passer ou d'organismes extérieurs, sauf comme à la fin des processus sont gauche dans leur état initial. | " |
L'état initial de l'organisme, selon Gibbs, est supposée être telle que "le corps peut être amené à passer de là à des états de énergie dissipée par processus réversibles ". Dans son 1876 œuvre maîtresse Sur l'équilibre des substances hétérogènes, une analyse graphique des systèmes chimiques multi-phases, il se est engagé ses réflexions sur l'énergie libre chimique en plein.
Définitions

L'énergie libre de Gibbs est défini comme:
qui est la même que:
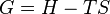
où:
- U est la énergie interne ( Unité SI: joule)
- p est égal à pression (unité SI: Pascal)
- V est le volume (unité SI: m 3)
- T est la température (unité SI: kelvin )
- S est la entropie (unité SI: joule par kelvin)
- H est l' enthalpie (unité SI: joule)
L'expression de la variation réversible infinitésimale dans l'énergie libre de Gibbs, pour un système ouvert, soumis à l'action des forces externes X i, qui provoquent des paramètres externes du système a i à changer d'une valeur da i, est donnée par:
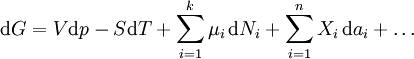
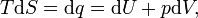 où:
où:
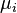 est le le potentiel chimique du i -ième composant chimique. (Unité SI: joules par particule ou joules par mole)
est le le potentiel chimique du i -ième composant chimique. (Unité SI: joules par particule ou joules par mole) 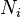 est le nombre de particules (ou nombre de moles) composant le composant chimique de la i.
est le nombre de particules (ou nombre de moles) composant le composant chimique de la i.
Ce est une forme de l'équation fondamentale Gibbs. Dans l'expression infinitésimale, le terme impliquant les comptes potentiels chimiques des changements dans énergie libre de Gibbs résultant d'un afflux de particules ou de flux sortant. En d'autres termes, elle est valable pour un système ouvert. Pour un système fermé, ce terme peut être abandonnée.
Ne importe quel nombre de termes supplémentaires peuvent être ajoutées, selon le système particulier considéré. Mis à part le travail mécanique , un système peut en outre effectuer de nombreux autres types de travail. Par exemple, dans l'expression infinitésimale, l'énergie de travail de contraction associée à un système thermodynamique qui est une fibre contractile qui réduit d'une quantité - dl sous une force f se traduirait par un FDL terme étant ajouté. Si une quantité de charge - de se acquiert par un système à une Ψ potentiel électrique, les travaux d'électricité associée à ce est de -Ψ, qui seraient inclus dans l'expression infinitésimale. Autres conditions de travail sont ajoutés selon les exigences du système.
Chaque quantité dans les équations ci-dessus peut être divisée par la quantité de substance mesurée dans taupes, pour former molaire énergie libre Gibbs. L'énergie libre de Gibbs est l'une des fonctions thermodynamiques les plus importants pour la caractérisation d'un système. Ce est un facteur déterminant dans les résultats tels que la une tension de cellule électrochimique, et la une constante d'équilibre de réaction réversible. Dans isotherme, systèmes isobariques, énergie libre de Gibbs peut être considéré comme une quantité «dynamique», en ce qu'elle est une mesure représentative des effets opposés des forces de enthalpiques et entropique conduite impliqués dans un processus thermodynamique.
La dépendance en température de l'énergie de Gibbs pour un gaz parfait est donnée par la Gibbs-Helmholtz équation et sa pression dépendance est donné par:
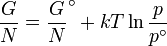
si le volume est connu plutôt que la pression il devient alors:
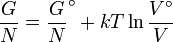
ou plus commodément que son potentiel chimique:
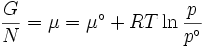
Dans les systèmes non-idéales, fugacité entre en jeu.
Dérivation
L'énergie libre de Gibbs différentielle totale en termes de variables naturelles peuvent être obtenues via Legendre transforme du énergie interne. Pour un système subissant un processus interne réversible qui est autorisé à échanger affaire, la chaleur et le travail avec son environnement, le différentiel de l'énergie interne est donnée de la première loi de la thermodynamique que
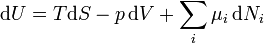 .
.
Parce que 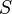 ,
, 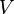 Et
Et 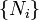 sont variables extensives, Fonction homogène théorème d'Euler permet une intégration facile des
sont variables extensives, Fonction homogène théorème d'Euler permet une intégration facile des 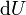 :
:
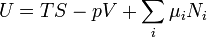 .
.
La définition de 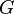 d'en haut est
d'en haut est
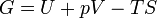 .
.
Prenant la différentielle totale, nous avons
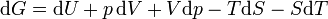 .
.
Remplacement avec le résultat de la première loi donne
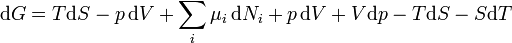
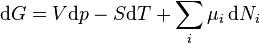 .
.
Les variables naturelles de sont alors 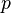 ,
,  Et
Et 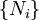 . Parce que certaines des variables naturelles sont intensifs,
. Parce que certaines des variables naturelles sont intensifs,  ne peuvent pas être intégrés à l'aide d'Euler intégrales comme ce est le cas avec l'énergie. Cependant, en remplaçant simplement le résultat pour
ne peuvent pas être intégrés à l'aide d'Euler intégrales comme ce est le cas avec l'énergie. Cependant, en remplaçant simplement le résultat pour  dans la définition de donne une expression standard pour G:
dans la définition de donne une expression standard pour G:
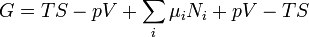
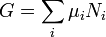 .
.
Vue d'ensemble
De manière simple, en ce qui concerne STP systèmes réagir, un général règle de base est:
| " | Chaque système vise à atteindre un minimum d'énergie libre. | " |
Par conséquent, en dehors de cette tendance naturelle général, une mesure quantitative de la façon dont près ou de loin est une réaction potentielle de ce minimum est calculé lors de l'énergétique du procédé indiquent que le changement de Gibbs Ag énergie libre est négatif. Essentiellement, cela signifie que cette réaction est favorisée et libérer de l'énergie. L'énergie libérée est égale à la quantité maximale de travail qui peut être effectuée à la suite de la réaction chimique. Inversement, si les conditions indiqué une Ag positive, alors l'énergie - sous la forme de travail - devrait être ajouté au système de réagir à rendre la réaction aller.
Histoire
La quantité appelée "énergie libre" est essentiellement un remplacement plus avancée et précise pour le terme obsolète " affinité », qui a été utilisé par les chimistes dans les années précédentes pour décrire la« force »qui a provoqué des réactions chimiques . Le terme d'affinité, tel qu'il est utilisé en relation chimique, remonte au moins au moment de Albertus Magnus en 1250.
De 1998 les manuels modernes Thermodynamique par le prix Nobel et professeur de génie chimique Ilya Prigogine nous trouvons:? "Comme motion a été expliqué par le concept newtonien de la force, les chimistes voulaient un concept similaire de« force motrice »pour changement chimique Pourquoi les réactions chimiques se produisent, et pourquoi se arrêtent pas aux certains points chimistes appelé le ' force »qui a provoqué des réactions chimiques affinité, mais il lui manquait une définition claire".
Au cours du 18ème siècle tout entier, l'opinion dominante en ce qui concerne la chaleur et de la lumière, ce est que mis en avant par Isaac Newton , appelé «l'hypothèse de Newton", qui a déclaré que la lumière et la chaleur sont les formes de matière attirés ou repoussés par d'autres formes de la matière, avec analogues aux forces de gravitation ou affinité chimique.
Au 19ème siècle, le chimiste français Marcellin Berthelot et le chimiste danois Julius Thomsen avait tenté de quantifier affinité en utilisant chaleurs de réaction. En 1875, après avoir quantifié les chaleurs de réaction pour un grand nombre de composés, Berthelot a proposé la " principe du travail maximal "dans lequel tous les changements chimiques qui se produisent sans intervention de l'énergie en dehors tendent vers la production de corps ou d'un système de corps qui libèrent la chaleur .
En plus de cela, en 1780 Antoine Lavoisier et Pierre-Simon Laplace ont jeté les bases de thermochimie en montrant que la chaleur dégagée dans une réaction est égale à la chaleur absorbée dans la réaction inverse. Ils ont également étudié la chaleur spécifique et chaleur latente d'un certain nombre de substances, et les quantités de chaleur dégagée lors de la combustion. De même, en 1840 chimiste suisse Germain Hess formulé le principe que le dégagement de chaleur dans une réaction est le même que le procédé est réalisé en une seule étape ou en plusieurs étapes. Ceci est connu comme La loi de Hess. Avec l'avènement de la la théorie mécanique de la chaleur au début du 19e siècle, la loi de Hess est venu à être considéré comme une conséquence de la loi de conservation de l'énergie.
Sur la base de ces idées, et d'autres Berthelot et Thomsen, ainsi que d'autres, examiné la chaleur dégagée dans la formation d'un composé comme une mesure de l'affinité, ou le travail effectué par les forces chimiques. Ce point de vue, cependant, ne était pas tout à fait correcte. En 1847, le physicien anglais James Joule a montré qu'il pouvait élever la température de l'eau en tournant une roue à aubes en elle, montrant ainsi que la chaleur et le travail mécanique étaient équivalentes ou proportionnelle à l'autre, soit environ, 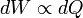 . Cette déclaration est venu à être connu sous le nom équivalent mécanique de la chaleur et était une forme précurseur de la première loi de la thermodynamique .
. Cette déclaration est venu à être connu sous le nom équivalent mécanique de la chaleur et était une forme précurseur de la première loi de la thermodynamique .
En 1865, le physicien allemand Rudolf Clausius avait montré que ce principe d'équivalence nécessaire amendement. Autrement dit, on peut utiliser la chaleur provenant d'un réaction de combustion dans un four à charbon pour faire bouillir l'eau, et d'utiliser cette chaleur pour vaporiser la vapeur, puis utiliser l'énergie à haute pression accrue de la vapeur vaporisé pour pousser un piston. Ainsi, nous pourrions naïvement raison que l'on peut tout à convertir la chaleur de combustion initiale de la réaction chimique dans le travail de pousser le piston. Clausius a montré, cependant, que nous devons prendre en compte le travail que les molécules de l'organe de travail, à savoir les molécules d'eau dans le cylindre, le faire sur l'autre lors de leur passage ou transformer d'une étape ou état de la cycle du moteur à l'autre, par exemple à partir de (P1, V1) à (P2, V2). Clausius origine appelé cela le «contenu de transformation" du corps, et puis plus tard a changé le nom de l'entropie . Ainsi, la chaleur utilisée pour transformer le corps de travail de molécules d'un état à l'autre ne peut pas être utilisé pour faire un travail externe, par exemple pour pousser le piston. Clausius défini cette chaleur de transformation que dQ = TDS.
En 1873, Willard Gibbs publié une méthode de représentation géométrique des propriétés thermodynamiques des substances au moyen de surfaces dans laquelle il a présenté le plan préliminaire des principes de sa nouvelle équation permettant de prédire ou estimer les tendances de divers processus naturels de se ensuivre quand les corps ou les systèmes sont mis en contact. En étudiant les interactions de substances homogènes en contact, ce est à dire les corps, étant dans la composition partie solide, partie liquide, et une partie de la vapeur, et en utilisant un trois dimensions du volume - entropie - graphe intérieur de l'énergie, Gibbs était en mesure de déterminer trois états d'équilibre, ce est à dire «nécessairement stable», «neutre» et «instable», et si oui ou non les changements se ensuivra. En 1876, Gibbs construit sur ce cadre en introduisant le concept de potentiel chimique afin de tenir compte des réactions chimiques et des états d'organes qui sont chimiquement différents de l'autre. Dans ses propres mots, pour résumer ses résultats en 1873, Gibbs déclare:
Dans cette description, tel qu'il est utilisé par Gibbs, ε se réfère à la l'énergie interne du corps, η se réfère à la entropie du corps, et ν est le le volume du corps.
Ainsi, en 1882, après l'introduction de ces arguments par Clausius et Gibbs, le scientifique allemand Helmholtz a indiqué, à l'encontre de l'hypothèse de Berthelot et Thomas que l'affinité chimique est une mesure de la chaleur de réaction de la réaction chimique basé sur le principe du travail maximal, que l'affinité ne est pas la chaleur dégagée dans la formation d'un composé mais elle est la plus grande quantité de travail qui peut être obtenu lorsque la réaction est réalisée de manière réversible, par exemple le travail dans une cellule électrique réversible. Le travail maximale est donc considérée comme la diminution de la liberté, ou disponible, l'énergie du système (énergie libre de Gibbs G à T = constante, P = constante ou Helmholtz énergie libre F à T = constante, V = constante), tandis que le chaleur dégagée est habituellement une mesure de la diminution de l'énergie totale du système ( Énergie interne). Ainsi, G ou F est la quantité d'énergie «gratuite» pour les travaux dans les conditions données.
Jusqu'à ce point, l'avis général a été telle que: "toutes les réactions chimiques en voiture le système à un état d'équilibre dans lequel les affinités des réactions disparaissent". Au cours des 60 prochaines années, l'affinité terme est venu pour être remplacé par l'énergie libre terme. Selon l'historien de la chimie Henry Leicester, l'influent 1923 manuel Thermodynamique et l'énergie libre de réactions chimiques par Gilbert N. Lewis et Merle Randall a conduit au remplacement du terme «affinité» par le terme «énergie libre» dans une grande partie du monde anglo-saxon.
Que signifie le terme «libre» signifie?
Dans les 18e et 19e siècles, le théorie de la chaleur, ce est à dire que la chaleur est une forme d'énergie ayant rapport à un mouvement vibratoire, commençait à supplanter la fois le théorie calorique, ce est à dire que la chaleur est un fluide, et la quatre théorie de l'élément dans lequel la chaleur est le plus léger des quatre éléments. Beaucoup de manuels scolaires et des articles d'enseignement au cours de ces siècles présentés ces théories côte à côte. De même, au cours de ces années, la chaleur commençait à se distinguer en différentes catégories de classification, tels que, "production combinée de chaleur" "la chaleur libre», «la chaleur rayonnante», chaleur spécifique, la capacité de la chaleur, "la chaleur absolue", "calorique latent», «libre» ou «perceptible» calorique (calorique sensible), entre autres.
En 1780, par exemple, Laplace et Lavoisier a déclaré: «En général, on peut changer la première hypothèse dans la seconde en changeant les mots de la chaleur libre, production combinée de chaleur et chaleur libérée» en « . vis viva, perte de force vive, et l'augmentation de la force vive »De cette manière, la masse totale de calories dans un corps, appelé la chaleur absolue, a été considéré comme un mélange de deux composants; le calorique libre ou perceptible pourrait affecter un thermomètre tandis que l'autre composante, le calorique latent, ne pouvait pas. L'utilisation des mots "chaleur latente" impliquait une similitude à la chaleur latente dans le sens le plus courant; il a été considéré comme lié chimiquement aux molécules de l'organisme. Dans le adiabatique compression d'un gaz la chaleur absolue est restée constante par la hausse de la température observée, indiquant que certains calorique latente était devenu «libre» ou perceptible.
Au début du 19ème siècle, le concept de calorique perceptible ou libre a commencé à être appelé «la chaleur libre» ou chaleur dégagée. En 1824, par exemple, le physicien français Sadi Carnot, dans ses célèbres «Réflexions sur la puissance motrice du feu", parle de quantités de chaleur 'absorbée ou libérés »dans différentes transformations. En 1882, le physicien et physiologiste allemand Hermann von Helmholtz a inventé l'expression «énergie libre» pour l'expression E - TS, dans lequel le changement de F (ou G) détermine la quantité d' énergie «gratuite» pour les travaux dans les conditions données.
Ainsi, dans l'utilisation traditionnelle, le terme «libre» a été fixé à énergie libre de Gibbs, ce est à dire pour les systèmes à pression et température constante, ou Helmholtz énergie libre, ce est à dire pour les systèmes à volume constant et la température, pour signifier «disponibles sous la forme de un travail utile. » En référence à l'énergie libre de Gibbs, nous ajoutons la qualification que ce est l'énergie libre pour le travail non-volume.
Un nombre croissant de livres et articles de journaux ne incluent pas la pièce jointe "libre", se référant à G comme simplement de l'énergie Gibbs (et de même pour le L'énergie de Helmholtz). Ceci est le résultat d'un 1988 Réunion UICPA pour régler terminologies unifiées pour la communauté scientifique internationale, dans laquelle l'adjectif «libre» aurait été banni. Cette norme, toutefois, n'a pas encore été universellement adopté, et de nombreux articles publiés et les livres comprennent toujours le descriptif «libre».
Énergie libre de réactions
Pour calculer l'équation de l'énergie libre de Gibbs pour une système isolé, laissez S tot être l'entropie totale de la système isolé, ce est un système qui ne peut pas échanger de la chaleur ou de masse avec son environnement. Selon la seconde loi de la thermodynamique :

et si 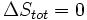 alors le processus est réversible. Le transfert de chaleur
alors le processus est réversible. Le transfert de chaleur 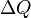 disparaît pour un système adiabatique. Tout processus adiabatique qui est également réversible est appelé isentropique
disparaît pour un système adiabatique. Tout processus adiabatique qui est également réversible est appelé isentropique 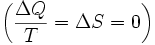 processus.
processus.
Considérons maintenant les systèmes diabatiques, ayant interne entropie S int. Un tel système est relié thermiquement à son environnement, qui ont entropie S ext. La forme d'entropie de la deuxième loi ne se applique pas directement au système diabatique, elle se applique uniquement au système fermé formé par le système et ses environs. Par conséquent, un processus est possible si
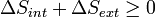 .
.
Nous allons essayer d'exprimer le côté gauche de cette équation entièrement en termes de fonctions de l'État ΔS poste est défini comme.:
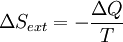
La température T est la même pour deux systèmes en équilibre thermique. Par le Principe zéro de la thermodynamique, si un système est en équilibre thermique avec un second et un troisième système, ces deux derniers sont en équilibre aussi bien. Aussi, est la chaleur transférée au système, de sorte que 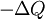 est la chaleur transférée à l'environnement, et -ΔQ / T est acquise par entropie les environs. Nous avons maintenant:
est la chaleur transférée à l'environnement, et -ΔQ / T est acquise par entropie les environs. Nous avons maintenant:
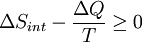
Multipliez les deux côtés par T:
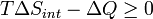
AQ est la chaleur transférée vers le système; si le processus est maintenant supposé être isobarique, puis AQ p = AH:
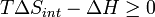
AH est la variation d'enthalpie de la réaction (pour une réaction chimique à pression constante et de la température). Puis
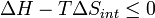
pour un processus possible. Laissez le changement Δ G énergie libre de Gibbs être définie comme
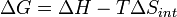 (Eq.1)
(Eq.1)
Notez qu'il ne est pas défini en termes de toutes les fonctions de l'État externes, tels que Δ S poste ou Δ S tot. Puis la seconde loi devient, qui nous parle aussi de la spontanéité de la réaction:
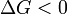 réaction favorisée (spontanée)
réaction favorisée (spontanée) 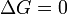 Ni l'avant ni la réaction inverse prévaut ( Equilibrium )
Ni l'avant ni la réaction inverse prévaut ( Equilibrium ) 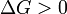 réaction défavorisée (Nonspontaneous)
réaction défavorisée (Nonspontaneous)
Énergie libre de Gibbs G se est défini comme
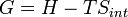 (Eq.2)
(Eq.2)
Mais remarquez que, pour obtenir l'équation (2) de l'équation (1) nous devons supposer que T est constante. Ainsi, énergie libre de Gibbs est très utile pour des procédés thermochimiques à température constante et la pression: les deux isothermes et isobares. Ces processus ne se déplacent pas sur un P - T diagramme, tels que le changement de phase d'un corps pur, qui se déroule à la pression de saturation et la température. Les réactions chimiques, cependant, ne subissent des changements dans le potentiel chimique, qui est une fonction d'état. Ainsi, les processus thermodynamiques ne se limitent pas aux deux dimensions P - V diagramme. Il existe une troisième dimension de n, la quantité de gaz. Naturellement pour l'étude des produits chimiques explosifs, les procédés ne sont pas nécessairement isotherme et isobare. Pour ces études, Énergie libre est utilisé.
Si un système fermé (Δ Q = 0) est à pression constante (Δ Q = Δ H), puis
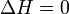
Par conséquent, l'énergie libre d'un système fermé est:
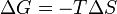
et si 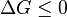 alors cela signifie que
alors cela signifie que 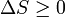 , À l'endroit où nous avons commencé la dérivation de Δ G.
, À l'endroit où nous avons commencé la dérivation de Δ G.
Identités utiles
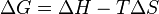 à température constante
à température constante 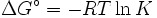
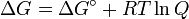
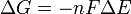
et réarrangement donne
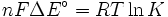
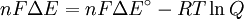
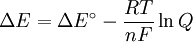
qui concerne le potentiel électrique d'une réaction au coefficient d'équilibre de cette réaction.
où
Δ G = changement énergie libre de Gibbs, Δ H = changement dans enthalpie , T = absolue température , Δ S = changement de l'entropie , R = constante des gaz, ln = logarithme naturel , K = la constante d'équilibre, Q = quotient de réaction, n = nombre d' électrons par produit taupe, F = Constante de Faraday ( coulombs par mole), et Δ E = le potentiel électrique de la réaction. En outre, nous avons aussi:
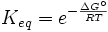
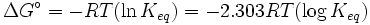
qui concerne l'équilibre constant avec énergie libre de Gibbs.
Changement standard de formation
L'énergie libre de Gibbs norme de formation d'un composé est le changement de l'énergie libre de Gibbs qui accompagne la formation d'une mole de cette substance à partir de ses éléments constitutifs, à leur états standards (la forme la plus stable de l'élément à 25 degrés Celsius et 100 kilopascals). Son symbole est Δ G f O.
Tous les éléments dans leurs états standard ( oxygène , gaz graphite, etc.) ont 0 changement d'énergie norme Gibbs libre de formation, car il n'y a pas de changement en cause.
- Δ G = Δ G ˚ + RT ln Q
A l'équilibre, Δ G = 0 et Q = K donc l'équation devient Δ G ˚ = - RT ln K
Table des matières sélectionnés
| Substance | État | Δ G ˚ ( cal / mol) |
|---|---|---|
| NH 3 | g | -3,976 |
| H 2 O | lq | -56,69 |
| H 2 O | g | -54,64 |
| CO 2 | g | -94,26 |
| CO | g | -32,81 |
| CH 4 | g | -12,14 |
| C 2 H 6 | g | -7,86 |
| C 3 H 8 | g | -5,614 |
| C 8 H 18 | g | 4.14 |
| C 10 H 22 | g | 8,23 |