
Química quântica
Você sabia ...
Crianças SOS tentou tornar o conteúdo mais acessível Wikipedia por esta selecção escolas. Patrocinar uma criança para fazer uma diferença real.
Química quântica é um ramo da química teórica, que se aplica a mecânica quântica e teoria quântica de campos para tratar de questões e problemas em química . A descrição da eletrônica comportamento dos átomos e moléculas como pertencente a sua reactividade é uma das aplicações de química quântica. Química quântica encontra-se na fronteira entre a química ea física , e as contribuições significativas foram feitas por cientistas de ambos os campos. Ele tem uma sobreposição forte e ativo com o campo da física atômica e física molecular , bem como química física.
Química quântica descreve matematicamente o comportamento fundamental da matéria no molecular escala. É, em princípio, possível descrever todos os sistemas químicos que utilizam esta teoria. Na prática, só os sistemas químicos mais simples podem realisticamente ser investigada em puramente mecânica quântica termos, e aproximações devem ser feitas para fins mais práticos (por exemplo, Hartree-Fock, pós Hartree-Fock ou Teoria do funcional da densidade, consulte química computacional para mais detalhes). Assim um entendimento detalhado de mecânica quântica não é necessário para a maioria química, como as implicações importantes da teoria (principalmente o aproximação orbital) pode ser entendida e aplicada em termos mais simples.
Na mecânica quântica (várias aplicações em química computacional e química quântica), o Hamiltoniano, ou o estado físico, de uma partícula pode ser expressa como a soma de dois operadores, um correspondendo a energia cinética e a outra para energia potencial. O Hamiltonian na equação de onda de Schrödinger utilizado em química quântica não contém termos para a rotação do elétron.
Soluções da equação de Schrödinger para o átomo de hidrogénio dá a forma de onda para a função orbitais atómicas, ea energia relativa das várias orbitais. A aproximação da órbita podem ser utilizados para compreender outros átomos, por exemplo, hélio , lítio e carbono .
História
A história da química quântica, essencialmente, iniciou-se com a descoberta de 1838 raios catódicos por Michael Faraday , a declaração de 1859 problema radiação de corpo negro por Gustav Kirchhoff, a sugestão de 1877 Ludwig Boltzmann que os estados de energia de um sistema físico podia ser discreta, e o 1900 hipótese quântica por Max Planck que qualquer energia radiante sistema atómica pode, teoricamente, ser dividido em um certo número de elementos discretos de energia ε tal que cada um destes elementos de energia é proporcional o ν frequência com a qual cada um deles individualmente irradiar energia , tal como definido pela seguinte fórmula:
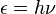
em que h é um valor numérico chamada Constante de Planck. Em seguida, em 1905, para explicar o efeito fotoelétrico (1839), ou seja, aquela luz brilhante em determinados materiais podem funcionar para ejetar elétrons do material, Albert Einstein postulou, com base na hipótese quântica de Planck, que a luz em si consiste de partículas quânticas individuais, que mais tarde veio a ser chamado de fótons ( 1926). Nos anos que se seguem, esta base teórica começou lentamente a ser aplicada a estrutura química, a reactividade, e ligação.
Estrutura eletrônica
O primeiro passo para resolver um problema de química quântica é normalmente resolver o Equação de Schrödinger (ou Equação de Dirac em química quântica relativística) com o Hamiltonian molecular eletrônico. Isto é chamado a determinação da estrutura electrónica da molécula. Pode-se dizer que a estrutura electrónica de uma molécula ou de cristal implica essencialmente as suas propriedades químicas.
Modelo de ondas
A fundação da mecânica quântica e química quântica é o modelo de onda, em que o átomo é uma pequena densa, carregada positivamente núcleo cercado por elétrons. Ao contrário do anteriormente Modelo de Bohr do átomo, no entanto, o modelo de onda descreve como elétrons " "nuvens que se deslocam em orbitais, e as suas posições são representadas pela distribuições de probabilidades em vez de pontos discretos. A força deste modelo reside na sua poder preditivo. Especificamente, ele prediz o padrão de elementos quimicamente semelhantes encontrados na tabela periódica . O modelo de onda é assim chamado porque os elétrons exibem propriedades (como interferência) tradicionalmente associada com as ondas. Veja dualidade onda-partícula .
Valence vínculo
Embora a base matemática de química quântica tinha sido colocado por Schrödinger em 1926 , é geralmente aceite que o primeiro verdadeiro cálculo em química quântica foi a dos físicos alemães Walter e Heitler Fritz Londres sobre o hidrogênio (H 2) molécula em 1927 . O método de Heitler e Londres foi prorrogado pelo físico teórico americano John C. Slater eo químico teórico americano Linus Pauling para se tornar o Valence-Bond (VB) [ou Heitler-Londres-Slater-Pauling (HLSP)] método. Neste método, a atenção é principalmente dedicado às interações de pares entre átomos e, portanto, este método correlaciona-se estreitamente com desenhos de 'químicos clássicos títulos .
Orbital molecular
Uma abordagem alternativa foi desenvolvida em 1929 por Friedrich Hund e Robert S. Mulliken, no qual os elétrons são descritos por funções matemáticas deslocalizada sobre toda uma molécula . A abordagem Hund-Mulliken ou método molecular orbital (MO) é menos intuitiva para os químicos, mas acabou capaz de predizer propriedades espectroscópicas melhor do que o método VB. Esta aproximação é a base da concepcional Método Hartree-Fock e mais métodos pós Hartree-Fock.
Teoria do funcional da densidade
O Thomas-Fermi modelo foi desenvolvido de forma independente pela Thomas e Fermi, em 1927 . Esta foi a primeira tentativa de descrever sistemas de muitos elétrons com base em densidade electrónica em vez de As funções de onda, embora não tenha sido muito bem sucedida no tratamento de moléculas inteiras. O método não fornecer a base para o que hoje é conhecido como teoria do funcional da densidade. Embora este método é menos desenvolvida do que os métodos pós Hartree-Fock, os seus requisitos computacionais menores lhe permitirão resolver os maiores moléculas poliatómicos e até mesmo macromoléculas, o que tornou o método mais utilizado em química computacional no momento.
Chemical Dynamics
Um passo adicional pode consistir de resolver o Equação de Schrödinger com o total Hamiltoniano molecular, a fim de estudar o movimento das moléculas. Solução direta da equação de Schrödinger é chamado de dinâmica molecular quântica, dentro do aproximação semiclássica dinâmica molecular semiclássicos, e dentro da mecânica clássica quadro dinâmica molecular (MD). Abordagens estatísticas, utilizando, por exemplo, métodos de Monte Carlo , também são possíveis.
Dinâmica química adiabáticos
Na dinâmica adiabática, interações interatômicas são representados por único escalar potenciais chamado superfícies de energia potencial. Isto é o Born-Oppenheimer aproximação introduzido por Nascido e Oppenheimer em 1927 . Aplicações pioneiras deste em química foram realizadas por Arroz e Ramsperger em 1927 e Kassel em 1928 , e generalizada no Teoria RRKM em 1952 por Marcus que tomou a teoria do estado de transição desenvolvido por Eyring em 1935 em consideração. Estes métodos permitem estimativas simples de unimoleculares taxas de reacção de algumas características da superfície potencial.
Dinâmica química não-adiabáticos
Dinâmica não-adiabáticos consiste em tomar a interação entre vários superfície de energia potencial acoplado (correspondendo a diferentes eletrônico estados quânticos da molécula). Os termos de acoplamento são chamados acoplamentos vibrônicos. O trabalho pioneiro neste campo foi feito por Stueckelberg, Landau, e Zener nos anos 1930 , em seu trabalho sobre o que agora é conhecido como o Transição de Landau-Zener. Sua fórmula permite que a probabilidade de transição entre dois curvas diabático potenciais no bairro de um cruzamento evitado a ser calculado.
Química quântica e teoria quântica de campos
A aplicação da teoria quântica de campos (QFT) para sistemas químicos e teorias tornou-se cada vez mais comum nas modernas ciências físicas. Um dos primeiros e mais fundamentalmente explícitas aparências deste é visto na teoria da photomagneton. Neste sistema, plasmas , que são onipresentes em ambos física e química, são estudadas a fim de determinar a base quantização do subjacente bosônico campo. No entanto, a teoria quântica de campos é de interesse em muitos campos da química, incluindo: química nuclear, astrochemistry, Sonochemistry, e hidrodinâmica quântica. Campo métodos teóricos também têm sido essenciais no desenvolvimento do ab initio eficaz Hamiltonian teoria dos métodos de pi-elétron semi-empíricos.