

Chimie quantique
Saviez-vous ...
SOS Enfants a essayé de rendre le contenu plus accessible Wikipedia par cette sélection des écoles. Parrainer un enfant de faire une réelle différence.
Chimie quantique est une branche de chimie théorique, qui se applique la mécanique quantique et la théorie quantique des champs pour aborder les questions et les problèmes de la chimie . La description de l' électronique comportement des atomes et des molécules comme se rapportant à leur réactivité est l'une des applications de la chimie quantique. Chimie quantique se trouve sur la frontière entre la chimie et la physique , et d'importantes contributions ont été faites par des scientifiques de deux champs. Il a un chevauchement forte et active avec le domaine de la physique atomique et physique moléculaire , ainsi que chimie physique.
Chimie quantique décrit mathématiquement le comportement fondamental de la matière à l' moléculaire échelle. Il est, en principe, possible de décrire tous les systèmes chimiques à l'aide de cette théorie. Dans la pratique, seuls les systèmes chimiques simples peuvent être étudiés dans réaliste purement mécanique quantique termes et approximations doivent être faites à des fins plus pratiques (par exemple, Hartree-Fock, après Hartree-Fock ou Densité de théorie de la fonctionnelle, voir la chimie computationnelle pour plus de détails). Ainsi une compréhension détaillée de la mécanique quantique ne est pas nécessaire pour la plupart la chimie, que les implications importantes de la théorie (principalement le rapprochement orbitale) peut être comprise et appliquée en termes plus simples.
En mécanique quantique (plusieurs applications en chimie computationnelle et de la chimie quantique), le Hamiltonien, ou l'état physique, d'une particule peut être exprimée comme la somme de deux opérateurs, l'un correspondant à l'énergie cinétique et l'autre à énergie potentielle. Le Hamiltonien dans l' équation d'onde de Schrödinger utilisé en chimie quantique ne contient pas de termes pour le spin de l'électron.
Les solutions de l'équation de Schrödinger pour l'atome d'hydrogène donne la forme de la fonction d'onde pour orbitales atomiques, et l'énergie relative des différentes orbitales. L'approximation orbital peut être utilisé pour comprendre les autres atomes, par exemple de l'hélium , de lithium et de carbone .
Histoire
L'histoire de la chimie quantique essentiellement a commencé avec la découverte de 1838 des rayons cathodiques par Michael Faraday , la déclaration de la 1859 problème de rayonnement du corps noir par Gustav Kirchhoff, la suggestion de 1877 Ludwig Boltzmann ce que les états d'énergie d'un système physique pourrait être discret, et le 1900 hypothèse quantique par Max Planck que toute l'énergie rayonnante système atomique peut théoriquement être divisée en un certain nombre d'éléments discrets d'énergie ε de telle sorte que chacun de ces éléments d'énergie est proportionnelle à la ν fréquence avec laquelle ils émettent chacun individuellement l'énergie , telle que définie par la formule suivante:
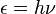
où h est une valeur numérique appelée La constante de Planck. Puis, en 1905, pour expliquer la effet photoélectrique (1839), ce est à dire, que la lumière sur certains matériaux brille peut fonctionner pour éjecter les électrons de la matière, Albert Einstein a postulé, sur la base de l'hypothèse quantique de Planck, que la lumière se compose de particules quantiques individuelles, qui sont venus plus tard appelé photons ( 1926). Dans les années suivantes, cette base théorique a lentement commencé à être appliqué à la structure chimique, la réactivité, et le collage.
Structure électronique
La première étape dans la résolution d'un problème chimique quantique est habituellement de résoudre le Équation de Schrödinger (ou Équation de Dirac dans relativiste chimie quantique) avec le hamiltonien électronique moléculaire. Ceci est appelé la détermination de la structure électronique de la molécule. On peut dire que la structure électronique d'une molécule de cristal ou implique essentiellement ses propriétés chimiques.
Vague modèle
Le fondement de la mécanique quantique et la chimie quantique est le modèle d'onde, dans lequel l'atome est un petit, dense, chargé positivement noyau entouré d'électrons. Contrairement à la précédente Modèle de Bohr de l'atome, cependant, le modèle d'onde des électrons décrit comme " nuages "se déplaçant dans orbitales et leurs positions sont représentées par distributions de probabilités plutôt que des points discrets. La force de ce modèle réside dans sa pouvoir prédictif. Plus précisément, on prévoit que le motif d'éléments chimiquement similaires trouvés dans la table périodique . Le modèle d'onde est ainsi nommé parce que les propriétés des électrons d'exposition (tels que les interférences) traditionnellement associés avec des vagues. Voir la dualité onde-particule .
Obligataire Valence
Bien que la base mathématique de la chimie quantique avait été posée par Schrödinger en 1926 , il est généralement admis que le premier véritable calcul en chimie quantique, ce est que des physiciens allemands Walter et Heitler Fritz Londres le hydrogène (H 2) molécule dans 1927 . La méthode de Heitler et Londres a été prolongée par le physicien théoricien américain John C. Slater et le chimiste théoricien américain Linus Pauling de devenir le Valence-Bond (VB) [ou Heitler-Londres-Slater-Pauling (HLSP)] méthode. Dans cette méthode, l'attention est principalement consacré aux interactions par paire entre atomes, et cette méthode corrèle donc étroitement avec les dessins de chimistes classiques de liaisons .
Orbitale moléculaire
Une autre approche a été développée en 1929 par Friedrich Hund et Robert Mulliken, dans lequel les électrons sont décrits par des fonctions mathématiques délocalisée sur toute une molécule . L'approche Hund-Mulliken ou la méthode des orbitales moléculaires (MO) est moins intuitif pour les chimistes, mais se est avéré capable de prédire les propriétés spectroscopiques mieux que la méthode de VB. Cette approche est la base conceptuelle de la Méthode Hartree-Fock et plus méthodes de post Hartree-Fock.
théorie de la fonctionnelle de la densité
Le Modèle de Thomas-Fermi a été développé indépendamment par Thomas et Fermi dans 1927 . Ce était la première tentative de décrire les systèmes à plusieurs électrons sur la base de densité électronique au lieu de fonctions d'onde, même si elle ne était pas très efficaces dans le traitement de molécules entières. La méthode a fourni la base de ce qui est maintenant connu comme théorie de la fonctionnelle de la densité. Bien que cette méthode est moins développé que soumettre méthodes Hartree-Fock, ses exigences de calcul inférieurs permettent de se attaquer à de plus grandes molécules polyatomiques et même macromolécules, qui en a fait la méthode la plus utilisée dans la chimie computationnelle à l'heure actuelle.
la dynamique chimiques
Une étape supplémentaire peut consister en résolvant le Équation de Schrödinger avec le total hamiltonien moléculaire pour étudier le mouvement des molécules. Solution directe de l'équation de Schrödinger est appelé dynamique moléculaire quantique, dans le approximation semi-classique dynamique moléculaire semi-classiques, et dans la mécanique classique cadre dynamique moléculaire (MD). Approches statistiques, en utilisant par exemple des méthodes de Monte Carlo , sont également possibles.
Dynamique chimique adiabatique
En dynamique adiabatique, interactions entre atomes sont représentés par simple scalaire potentiels appelés surfaces d'énergie potentielle. Ceci est le Born-Oppenheimer rapprochement présenté par Né et Oppenheimer en 1927 . Applications pionnières de cette chimie ont été effectuées par Rice et Ramsperger en 1927 et Kassel en 1928 et généralisé dans le théorie RRKM en 1952 par Marcus qui a pris la théorie de l'état de transition élaboré par Eyring en 1935 en compte. Ces méthodes permettent des estimations simples de unimoléculaires taux de réaction de quelques caractéristiques de la surface de potentiel.
Dynamique chimique non adiabatique
Dynamique non adiabatique consiste à prendre l'interaction entre plusieurs surface d'énergie potentielle associée (correspondant à différent électronique états quantiques de la molécule). Les termes de couplage sont appelés couplages vibroniques. Le travail de pionnier dans ce domaine a été réalisée par Stueckelberg, Landau, et Zener dans les années 1930 , dans leur travail sur ce qui est maintenant connu sous le nom Landau-Zener transition. Leur formule permet la probabilité de transition entre deux diabatiques courbes de potentiel à proximité d'une croisement évité à calculer.
Chimie quantique et la théorie quantique des champs
L'application de la théorie quantique des champs (QFT) aux systèmes et théories chimiques est devenue de plus en plus courante dans les sciences physiques modernes. L'un des premiers et des plus fondamentalement explicites apparences de ce est vu dans la théorie de la photomagneton. Dans ce système, les plasmas , qui sont omniprésents dans la physique et la chimie, sont étudiés afin de déterminer la base quantification du sous-jacent champ de bosons. Cependant, la théorie quantique des champs est d'intérêt dans de nombreux domaines de la chimie, y compris: chimie nucléaire, astrochimie, sonochimie, et hydrodynamique quantique. Terrain méthodes théoriques ont également été critique dans le développement de la théorie des méthodes pi-électroniques semi-empiriques ab initio efficace hamiltonien.