- Charles Perrault – Barba azul.pdf
- Charles Perrault – Las hadas.pdf
- Charles Perrault – Piel de Asno.pdf
- Charles Perrault – Riquet-el-del-copete.pdf
- Copia de Pardo Bazan, Emilia – Cuentos de la tierra.pdf
- Edgar Allan Poe – MANUSCRITO HALLADO EN UNA BOTELLA.pdf
- Palma, Ricardo – Fatuidad Humana.PDF
- Palma, Ricardo – La Pinga Del Libertado.PDF
- Palma, Ricardo – Tradiciones Peruanas.lit
- Pardo Bazan, Emilia – Camarona, La.pdf
- Pardo Bazan, Emilia – Cerezas, Las.pdf
- Pardo Bazan, Emilia – Cuentos de Marineda.pdf
- Pardo Bazan, Emilia – Cuentos de Navidad y Ano Nuevo.pdf
- Pardo Bazan, Emilia – Cuentos de Navidades y Reyes.pdf
- Pardo Bazan, Emilia – Cuentos de la patria.pdf
- Pardo Bazan, Emilia – Cuentos de la tierra.pdf
- Pardo Bazan, Emilia – Cuentos del terruno.pdf
- Pardo Bazan, Emilia – Cuentos dramaticos.pdf
- Pardo Bazan, Emilia – Cuentos nuevos.pdf
- Pardo Bazan, Emilia – Cuentos sacroprofanos.zip
- Pardo Bazan, Emilia – Cuentos tragicos.pdf
- Pardo Bazan, Emilia – Desde Alla.PDF
- Pardo Bazan, Emilia – Desde Alla.html
- Pardo Bazan, Emilia – Diptico.pdf
- Pardo Bazan, Emilia – Hijo Del Alma .pdf
- Pardo Bazan, Emilia – Historias y cuentos de Galicia.pdf
- Pardo Bazan, Emilia – Infidelidad.pdf
- Pardo Bazan, Emilia – Interiores.pdf
- Pardo Bazan, Emilia – Madre Naturaleza, La.pdf
- Pardo Bazan, Emilia – Milagro del hermanuco, El.pdf
- Pardo Bazan, Emilia – Novia fiel, La.pdf
- Pardo Bazan, Emilia – Otros cuentos.pdf
- Pardo Bazan, Emilia – Pazos de Ulloa, Los.pdf
- Pardo Bazan, Emilia – Reconciliados.pdf
- Pardo Bazan, Emilia – Senor doctoral, El.zip
- Pellegrini, Carlos – El arte de la guerra.lit
- Pereda, Jose Maria – Sotileza.pdf
- Pereda, Jose Maria de – Penas arriba.pdf
- Perez Galdos, Benito – 7 de Julio _1876_.pdf
- Perez Galdos, Benito – Aita Tettauen _1904_.pdf
- Perez Galdos, Benito – Amadeo I _1910_.pdf
- Perez Galdos, Benito – Bailen _1873_.pdf
- Perez Galdos, Benito – Bodas reales _1900_.pdf
- Perez Galdos, Benito – Cadiz _1874_.pdf
- Perez Galdos, Benito – Canovas _1912_.pdf
- Perez Galdos, Benito – Carlos VI en la Rapita _1904_.pdf
- Perez Galdos, Benito – De Cartago a Sagunto _1911_.pdf
- Perez Galdos, Benito – De Onate a La Granja _1898_.pdf
- Perez Galdos, Benito – Dona Perfecta _1876_.pdf
- Perez Galdos, Benito – El 19 de Marzo y el 2 de Mayo _1873_.pdf
- Perez Galdos, Benito – El 19 de marzo y el 2 de Mayo.pdf
- Perez Galdos, Benito – El Grande Oriente _1876_.pdf
- Perez Galdos, Benito – El abuelo _1897_.pdf
- Perez Galdos, Benito – El amigo Manso _1882_.pdf
- Perez Galdos, Benito – El audaz, historia de un radical de antan
- Perez Galdos, Benito – El doctor Centeno (1883).txt
- Perez Galdos, Benito – El terror de 1824 _1877_.pdf
- Perez Galdos, Benito – Electra _1901_.pdf
- Perez Galdos, Benito – Espana sin Rey _1907_.pdf
- Perez Galdos, Benito – Espana tragica _1909_.pdf
- Perez Galdos, Benito – Fortunata y Jacinta (dos historias de cas
- Perez Galdos, Benito – Gerona _1874_.pdf
- Perez Galdos, Benito – Gloria _1876_.pdf
- Perez Galdos, Benito – Halma _1895_.pdf
- Perez Galdos, Benito – Juan Martin el Empecinado _1874_.pdf
- Perez Galdos, Benito – La Fontana de Oro _1867_.pdf
- Perez Galdos, Benito – La Primera Republica _1911_.pdf
- Perez Galdos, Benito – La Segunda Casaca _1876_.pdf
- Perez Galdos, Benito – La Sombra -1871-.html
- Perez Galdos, Benito – La Sombra _1870_.pdf
- Perez Galdos, Benito – La batalla de los Arapiles _1875_.pdf
- Perez Galdos, Benito – La campana del Maestrazgo _1899_.pdf
- Perez Galdos, Benito – La corte de Carlos IV _1873_.pdf
- Perez Galdos, Benito – La de San Quintin _1894_.pdf
- Perez Galdos, Benito – La de bringas (1884).txt
- Perez Galdos, Benito – La de los tristes destinos _1907_.pdf
- Perez Galdos, Benito – La desheredada _1881_.pdf
- Perez Galdos, Benito – La estafeta romantica _1899_.pdf
- Perez Galdos, Benito – La familia de Leon Roch (1878).txt
- Perez Galdos, Benito – La fiera _1896_.pdf
- Perez Galdos, Benito – La incognita _1888_.pdf
- Perez Galdos, Benito – La loca de la casa _1892_.pdf
- Perez Galdos, Benito – La novela en el tranvia (1871).txt
- Perez Galdos, Benito – La revolucion de Julio _1904_.pdf
- Perez Galdos, Benito – La sociedad presente como materia novelab
- Perez Galdos, Benito – La vuelta al mundo en la Numancia (1906)
- Perez Galdos, Benito – Las tormentas del 48 _1902_.pdf
- Perez Galdos, Benito – Lo prohibido _1884_.pdf
- Perez Galdos, Benito – Los Ayacuchos _1900_.pdf
- Perez Galdos, Benito – Los Cien Mil Hijos de San Luis _1877_.pdf
- Perez Galdos, Benito – Los duendes de la camarilla _1903_.pdf
- Perez Galdos, Benito – Luchana _1899_.pdf
- Perez Galdos, Benito – Marianela _1878_.pdf
- Perez Galdos, Benito – Memorias de un cortesano de 1815 (1875).d
- Perez Galdos, Benito – Mendizabal _1898_.pdf
- Perez Galdos, Benito – Miau [PDF].pdf
- Perez Galdos, Benito – Miau _1888_.pdf
- Perez Galdos, Benito – Misericordia _1897_.pdf
- Perez Galdos, Benito – Montes de Oca _1900_.pdf
- Perez Galdos, Benito – Napoleon en Chamartin _1874_.pdf
- Perez Galdos, Benito – Narvaez _1902_.pdf
- Perez Galdos, Benito – Nazarin.html
- Perez Galdos, Benito – O’ Donnell _1904_.pdf
- Perez Galdos, Benito – Prim _1906_.pdf
- Perez Galdos, Benito – Realidad _1888_.pdf
- Perez Galdos, Benito – Sonemos alma sonemos (1903).html
- Perez Galdos, Benito – Tormento (1884).txt
- Perez Galdos, Benito – Torquemada en el Purgatorio _1894_.pdf
- Perez Galdos, Benito – Torquemada en la cruz _1893_.pdf
- Perez Galdos, Benito – Torquemada y San Pedro _1895_.pdf
- Perez Galdos, Benito – Trafalgar (1873).pdf
- Perez Galdos, Benito – Tristana (1892).pdf
- Perez Galdos, Benito – Tristana _vocabulario basico_.pdf
- Perez Galdos, Benito – Un faccioso mas y algunos frailes menos (
- Perez Galdos, Benito – Un voluntario realista _1878_.pdf
- Perez Galdos, Benito – Vergara _1899_.pdf
- Perez Galdos, Benito – Voluntad _1895_.pdf
- Perez Galdos, Benito – Zaragoza _1874_.pdf
- Perez Galdos, Benito – Zumalacarregui _1898_.pdf
- Perez Zaragoza Godinez, Agustin – Dompareli Bocanegra.html
- Perrault, Charles – Caperucita Roja.pdf
- Perrault, Charles – Cenicienta.pdf
- Perrault, Charles – El gato con botas.pdf
- Perrault, Charles – Pulgarcito.pdf
- Petronio – La matrona de Efeso.pdf
- Pierce, Ambrose – El club de los parricidas.zip
- Pitagoras – Los Versos de Oro.PDF
- Platon – Apologia de Socrates.pdf
- Platon – Carmide.pdf
- Platon – Cratilo.pdf
- Platon – Criton.pdf
- Platon – El Banquete.pdf
- Platon – Eutidemo.pdf
- Platon – Eutifron.pdf
- Platon – Fedon.pdf
- Platon – Gorgias.pdf
- Platon – Hipias Mayor.pdf
- Platon – Hipias Menor.pdf
- Platon – Ion.pdf
- Platon – La Republica.pdf
- Platon – Laques.pdf
- Platon – Lisis.pdf
- Platon – Menexeno.pdf
- Platon – Protagoras.pdf
- Platon – Protagoras.txt
- Platon – Timeo.pdf
- Plotino – Sobre el bien o el uno.pdf
- Poe, Edgar Allan – Annabel Lee.pdf
- Poe, Edgar Allan – Aventuras de Arthur Gordon Pym.pdf
- Poe, Edgar Allan – Berenice.htm
- Poe, Edgar Allan – Caja oblonga, La.pdf
- Poe, Edgar Allan – Cita, La.pdf
- Poe, Edgar Allan – Conversacion de Eiros y Charmion.pdf
- Poe, Edgar Allan – Cottage landor.pdf
- Poe, Edgar Allan – Cuatro Bestias en Una, El Hombre Cameleopardo
- Poe, Edgar Allan – Demonio de la perversidad.pdf
- Poe, Edgar Allan – Descenso al Maelstr_224m.pdf
- Poe, Edgar Allan – EL BARRIL DE AMONTILLADO.pdf
- Poe, Edgar Allan – EL CORAZoN DELATOR.pdf
- Poe, Edgar Allan – EL RETRATO OVAL.pdf
- Poe, Edgar Allan – El Barril de Amontillado (1846).pdf
- Poe, Edgar Allan – El Corazon Delator (1843).pdf
- Poe, Edgar Allan – El Cottage de Landor.htm
- Poe, Edgar Allan – El Cuervo.pdf
- Poe, Edgar Allan – El Diablo en el Campanario.htm
- Poe, Edgar Allan – El Entierro Prematuro.pdf
- Poe, Edgar Allan – El Escabarajo de Oro.pdf
- Poe, Edgar Allan – El Escarabajo de Oro.pdf
- Poe, Edgar Allan – El Hombre de la Multitud.htm
- Poe, Edgar Allan – El Hundimiento de la Casa Usher 1839.pdf
- Poe, Edgar Allan – El Poder de las Palabras.pdf
- Poe, Edgar Allan – El Pozo y el Pedulo.PDF
- Poe, Edgar Allan – El Pozo y el Pendulo 1842.pdf
- Poe, Edgar Allan – El Retrato Ovalado.pdf
- Poe, Edgar Allan – El gato negro.pdf
- Poe, Edgar Allan – El hundimiento de la Casa Usher.PDF
- Poe, Edgar Allan – Esfinge.pdf
- Poe, Edgar Allan – Gato Negro.pdf
- Poe, Edgar Allan – Gato Negro.txt
- Poe, Edgar Allan – Historias Extraordinarias.zip
- Poe, Edgar Allan – Hop-Frog.pdf
- Poe, Edgar Allan – Incomparable aventura de un tal Hans Pfaall.p
- Poe, Edgar Allan – LOS CRiMENES DE LA RUE MORGE.pdf
- Poe, Edgar Allan – La Carta Robada.pdf
- Poe, Edgar Allan – La Ciencia Ficcion de Edgar Allan Poe.pdf
- Poe, Edgar Allan – La Durmiente.pdf
- Poe, Edgar Allan – La Mascara de la Muerte Roja.pdf
- Poe, Edgar Allan – La Mascara de la Muerte Roja.txt
- Poe, Edgar Allan – La Verdad sobre el caso del Senor Valdemar 18
- Poe, Edgar Allan – Las Campanas.pdf
- Poe, Edgar Allan – Leonora.pdf
- Poe, Edgar Allan – Ligeia.pdf
- Poe, Edgar Allan – Los Crimenes de la Rue Morgue.pdf
- Poe, Edgar Allan – Los Hechos en el Caso de M. Valdemar.pdf
- Poe, Edgar Allan – Manuscrito Hallado en una Botella.htm
- Poe, Edgar Allan – Metzengerstein.pdf
- Poe, Edgar Allan – Misterio de Marie Roget, El.pdf
- Poe, Edgar Allan – Morella.pdf
- Poe, Edgar Allan – Poemas.pdf
- Poe, Edgar Allan – Revelaciones.pdf
- Poe, Edgar Allan – Silencio.lit
- Poe, Edgar Allan – Silencio.pdf
- Poe, Edgar Allan – Un Sueno en un Sueno.pdf
- Poe, Edgar Allan – Una historia de las montanas Ragged.html
- Poe, Edgar Allan – William Wilson.htm
- Polibio de Megalopolis – Historia universal bajo la Republica Ro
- Polo, Marco – El Libro de Marco Polo.pdf
- Polo, Marco – Viajes.txt
- Potocki, Jan – Historia Del Comendador De Toralva.PDF
- Potocki, Jan – Manuscrito encontrado en Zaragoza.lit
- Prado, Pedro – la reina de rapanui.pdf
- Proudhon, Pierre – El principio Federativo.pdf
- Proudhon, Pierre – Que es la propiedad.pdf
- Proust, Marcel – 1 Por el camino de Swann.pdf
- Proust, Marcel – 2 A la sombra de las muchachas en flor.pdf
- Proust, Marcel – 3 El mundo de Guermantes.pdf
- Proust, Marcel – 4 Sodoma y Gomorra.pdf
- Proust, Marcel – 5 La Prisionera.pdf
- Proust, Marcel – 6 Albertina desaparecida.pdf
- Proust, Marcel – 7 La fugitiva.pdf
- Proust, Marcel – 8 El tiempo recobrado.pdf
- Proust, Marcel – En busca del tiempo perdido I.pdf
- Proust, Marcel – Estudio sobre Marcel Proust.pdf
- Pushkin, Alexander – Boris Godunov.pdf
- Pushkin, Alexander – El fabricante de ataudes.pdf
- Pushkin, Alexander – La hija del capitan.pdf
- Pushkin, Alexander – Tempestad de nieve, La.pdf
- Pushkin, Alexander – Un disparo memorable.pdf

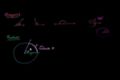 Unit circle definition of trig functions
Unit circle definition of trig functions Graphs of trig functions
Graphs of trig functions Trig identities and examples
Trig identities and examples Linear equations
Linear equations Linear inequalities
Linear inequalities Graphing and analyzing linear functions
Graphing and analyzing linear functions Systems of equations and inequalities
Systems of equations and inequalities Multiplying and factoring expressions
Multiplying and factoring expressions Quadratic equations
Quadratic equations Exponent expressions and equations
Exponent expressions and equations Functions
Functions Ratios and proportions
Ratios and proportions{kind=link}
-
Pages
- Adventure E-Books
- Afristory
- Audioletture
- Audiolibri
- Authors
- Autori
- Bible
- Biblioteca de Latino América
- Britannica
- Caruso
- Cat and Dog
- CD-ROM
- Christmas
- Conferenze
- Console
- Crux
- Devel
- Donazioni
- Dramatic
- EPUB
- For Schools
- Freeradius
- GNU
- Great Books
- Gutenberg
- Gutenberg Australia
- Hesperian Health
- Historical Fiction E-books
- Holy Bible
- Kaos
- KDE
- Liber Liber
- LiberMusica
- Libretti d’Opera
- Librivox
- Libro Parlato
- Linux
- Linux Mint
- Magnatune
- Malinverno
- Manuzio
- Marxists.org
- Old Software
- OpenOffice
- PG-072006
- PG-ISO
- PGP
- Processo per la morte di Giulio Regeni
- Public Domain Review Audio
- Public Domain Review Collection
- Rachmaninov: Complete Piano Concertos
- Requiem
- Standard
- Standard E-Books
- Textos
- Top 100 Favorites E-books
- Ubuntu
- Ubuntu
- Verdi
- Web
- Wikipedia 2006
- Wikipedia 2007
- Wikipedia 2008
- Wikipedia for Schools (EN)
- Wikipedia March 2008
- Women in African History
- WTC
Categories
- AAC
- Abba
- Abbate Migliore
- Abbott
- Accademia degli Intronati
- Accati
- Accetto
- Adams
- Africa
- Agamennone
- Aganoor
- Agaraff
- Agostini
- Agresti
- Agrippa
- Aho
- Ainslie
- Ainsworth
- Alamanni
- Alas
- Albergati Capacelli
- Albertazzi
- Alberti
- Albinoni
- Alcott
- Aleardi
- Algebra
- Alger
- Alighieri
- Alma Nova
- Almanova
- Altri Stromenti
- Amalteo
- Amari
- Amazon
- Anamar
- Andrea da Barberino
- Anonymous
- Appleton
- Aretino
- Ariosto
- Arnold
- Artusi
- Attivissimo
- Auber
- Audiobooks
- Audiolibri
- Austen
- Australia
- Authors
- Avolio
- azw3
- Baccini
- Bach
- Bachi
- Balbo
- Baldelli
- Balduzzi
- Balzac
- Barrili
- Beccaria
- Beier
- Bergonzi
- Bertelli
- Bhagavadgita
- Bible
- Biblioteca
- Blasco Ibáñez
- Boccaccio
- Boito
- Bonghi
- Booth
- Bortolotti
- Brahms
- Britannica
- Bronte
- Buonamento
- Burroughs
- Buxtehude
- Callas
- Canfora
- Cantini
- Canyar
- Cappato
- Caruso
- Castello
- Cavallini
- Cazzati
- CD-ROM
- Cecchini
- Challoner
- Chekhov
- Chopin
- Christmas
- Clarín
- Classical
- Collodi
- con Brio Choir
- Convegni
- Copyrighr
- Creative Commons
- Crux
- Danteide
- Davigo
- De Amicis
- De Falla
- De Sábata
- Debussy
- Deledda
- Della Casa
- Della Valle
- Di Fonzo
- Di Stefano
- Dickens
- Diritto d'autore
- Disney
- Donizetti
- DOS
- Dostoyevsky
- Douay
- Dowland
- Download
- Ebook
- Ebooks
- Edubuntu
- english
- EPUB
- Ethnical
- Fasol
- Fitch
- For Schools
- Francescatti
- Francesco d'Assisi
- Freeradius
- Gabrieli
- Galilei
- Garcia Lorca
- Garnett
- Giaches de Wert
- Giacosa
- Giampieretti
- GNU
- GNU/Linux
- GNUtemberg
- Goehr
- Goldberg
- Gospel
- Gramsci
- Grieg
- Guerrini
- Gutenberg
- Haydn
- Heringman
- Hesperian
- History
- HTML
- Ienna
- Illica
- In Nova Cantica
- Ishizaka
- iso
- Italia
- Johann C.
- Johnson
- Josquin des Prez
- Kammen
- Kaos
- KDE
- Kibbie
- Kindle
- King
- Kiwix
- Kobo
- La Reverie
- La Reveriei
- Latino-América
- Lawrence
- Lawson
- Legrenzi
- Lehner
- Leopardi
- Letteratura italiana
- Liber Liber
- Libretti
- Librivox
- Linux
- Lorenzini
- Luporini
- Lynne
- Macbeth
- Maggiora
- Magnatune
- Manzoni
- Martin
- Marx
- Marxists
- Mascagni
- Mattarella
- Matteo
- Medri
- Melli
- Meloni
- Melville
- Merula
- Milan
- Mitchell
- Monteverdi
- Montgomery
- Morales
- MP3
- Mudarra
- Mustrd Ebooks
- Nabucco
- Nazioni Unite
- Ockeghen
- Odoardi
- OGG
- ONU
- Open Source
- OpenOffice
- Opera
- Orlando di Lasso
- Ormandy
- Orwell
- Pachelbel
- Paganini
- Palazzolo
- Palestrina
- Paolone
- Paperback
- Patriquin
- Pavese
- Pentangle
- PGP
- Piave
- Piccinini
- Pienaar
- Pirandello
- Potevin
- Presidenza del Consiglio
- Project Gutenberg
- Public Domain
- Puccini
- Quevedo
- Rachmaninov
- Radio Radicale
- Raimondi
- Rambelli
- Ramin
- RAR
- Ravel
- Raymond
- Regeni
- Reusner
- Rheims
- Roncalli
- Ronco
- Rudel
- Rutter
- Saint-Exupéry
- Santanché
- Sardou
- Satie
- Scarlatti
- School
- SchoolLAN
- Schools
- Scopelliti
- Scores
- Sesse
- Sgrignoli
- Shakespeare
- Simonetto
- Simplicissimus Ensemble
- Software
- Soler
- Solidaria
- Spotify
- Spreaker
- St. John
- Stallman
- Standard Ebooks
- Stecchetti
- Storybooks
- Storytel
- Strozzi
- Suzzara Verdi
- tar
- Targioni Tozzetti
- Tarrega
- Telemann
- Teng
- Testa
- Textos
- Tolstoj
- Tortora
- Tortorelli
- Torvalds
- Toscanini
- Tozzi
- Trigonometry
- Trilussa
- Twain
- txt
- Ubuntu
- Uccellini
- Unamuno
- Uncategorized
- Unknown
- Valerio Di Stefano
- Vamba
- Vangelo
- Vannacci
- Vanzettii
- Verdi
- Verdinois
- Verga
- Verne
- Videos
- Vivaldi
- Voices of Music
- von Karajan
- Wahl
- Walnut Creek
- WAV
- Web
- Wells
- Wharton
- Wikipedia
- Wikipedia 2006
- Wilde
- Williams
- wim
- Windows
- WMA
- Women
- Woolf
- Youcanprint
- YouTube
- Zephyrus
- ZIM
- zip
- Zuncheddu
 The Voice in the Desert
The Voice in the Desert- Malinverno
Linux
Categories
- AAC
- Abba
- Abbate Migliore
- Abbott
- Accademia degli Intronati
- Accati
- Accetto
- Adams
- Africa
- Agamennone
- Aganoor
- Agaraff
- Agostini
- Agresti
- Agrippa
- Aho
- Ainslie
- Ainsworth
- Alamanni
- Alas
- Albergati Capacelli
- Albertazzi
- Alberti
- Albinoni
- Alcott
- Aleardi
- Algebra
- Alger
- Alighieri
- Alma Nova
- Almanova
- Altri Stromenti
- Amalteo
- Amari
- Amazon
- Anamar
- Andrea da Barberino
- Anonymous
- Appleton
- Aretino
- Ariosto
- Arnold
- Artusi
- Attivissimo
- Auber
- Audiobooks
- Audiolibri
- Austen
- Australia
- Authors
- Avolio
- azw3
- Baccini
- Bach
- Bachi
- Balbo
- Baldelli
- Balduzzi
- Balzac
- Barrili
- Beccaria
- Beier
- Bergonzi
- Bertelli
- Bhagavadgita
- Bible
- Biblioteca
- Blasco Ibáñez
- Boccaccio
- Boito
- Bonghi
- Booth
- Bortolotti
- Brahms
- Britannica
- Bronte
- Buonamento
- Burroughs
- Buxtehude
- Callas
- Canfora
- Cantini
- Canyar
- Cappato
- Caruso
- Castello
- Cavallini
- Cazzati
- CD-ROM
- Cecchini
- Challoner
- Chekhov
- Chopin
- Christmas
- Clarín
- Classical
- Collodi
- con Brio Choir
- Convegni
- Copyrighr
- Creative Commons
- Crux
- Danteide
- Davigo
- De Amicis
- De Falla
- De Sábata
- Debussy
- Deledda
- Della Casa
- Della Valle
- Di Fonzo
- Di Stefano
- Dickens
- Diritto d'autore
- Disney
- Donizetti
- DOS
- Dostoyevsky
- Douay
- Dowland
- Download
- Ebook
- Ebooks
- Edubuntu
- english
- EPUB
- Ethnical
- Fasol
- Fitch
- For Schools
- Francescatti
- Francesco d'Assisi
- Freeradius
- Gabrieli
- Galilei
- Garcia Lorca
- Garnett
- Giaches de Wert
- Giacosa
- Giampieretti
- GNU
- GNU/Linux
- GNUtemberg
- Goehr
- Goldberg
- Gospel
- Gramsci
- Grieg
- Guerrini
- Gutenberg
- Haydn
- Heringman
- Hesperian
- History
- HTML
- Ienna
- Illica
- In Nova Cantica
- Ishizaka
- iso
- Italia
- Johann C.
- Johnson
- Josquin des Prez
- Kammen
- Kaos
- KDE
- Kibbie
- Kindle
- King
- Kiwix
- Kobo
- La Reverie
- La Reveriei
- Latino-América
- Lawrence
- Lawson
- Legrenzi
- Lehner
- Leopardi
- Letteratura italiana
- Liber Liber
- Libretti
- Librivox
- Linux
- Lorenzini
- Luporini
- Lynne
- Macbeth
- Maggiora
- Magnatune
- Manzoni
- Martin
- Marx
- Marxists
- Mascagni
- Mattarella
- Matteo
- Medri
- Melli
- Meloni
- Melville
- Merula
- Milan
- Mitchell
- Monteverdi
- Montgomery
- Morales
- MP3
- Mudarra
- Mustrd Ebooks
- Nabucco
- Nazioni Unite
- Ockeghen
- Odoardi
- OGG
- ONU
- Open Source
- OpenOffice
- Opera
- Orlando di Lasso
- Ormandy
- Orwell
- Pachelbel
- Paganini
- Palazzolo
- Palestrina
- Paolone
- Paperback
- Patriquin
- Pavese
- Pentangle
- PGP
- Piave
- Piccinini
- Pienaar
- Pirandello
- Potevin
- Presidenza del Consiglio
- Project Gutenberg
- Public Domain
- Puccini
- Quevedo
- Rachmaninov
- Radio Radicale
- Raimondi
- Rambelli
- Ramin
- RAR
- Ravel
- Raymond
- Regeni
- Reusner
- Rheims
- Roncalli
- Ronco
- Rudel
- Rutter
- Saint-Exupéry
- Santanché
- Sardou
- Satie
- Scarlatti
- School
- SchoolLAN
- Schools
- Scopelliti
- Scores
- Sesse
- Sgrignoli
- Shakespeare
- Simonetto
- Simplicissimus Ensemble
- Software
- Soler
- Solidaria
- Spotify
- Spreaker
- St. John
- Stallman
- Standard Ebooks
- Stecchetti
- Storybooks
- Storytel
- Strozzi
- Suzzara Verdi
- tar
- Targioni Tozzetti
- Tarrega
- Telemann
- Teng
- Testa
- Textos
- Tolstoj
- Tortora
- Tortorelli
- Torvalds
- Toscanini
- Tozzi
- Trigonometry
- Trilussa
- Twain
- txt
- Ubuntu
- Uccellini
- Unamuno
- Uncategorized
- Unknown
- Valerio Di Stefano
- Vamba
- Vangelo
- Vannacci
- Vanzettii
- Verdi
- Verdinois
- Verga
- Verne
- Videos
- Vivaldi
- Voices of Music
- von Karajan
- Wahl
- Walnut Creek
- WAV
- Web
- Wells
- Wharton
- Wikipedia
- Wikipedia 2006
- Wilde
- Williams
- wim
- Windows
- WMA
- Women
- Woolf
- Youcanprint
- YouTube
- Zephyrus
- ZIM
- zip
- Zuncheddu
May 2024 M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 Encyclopaedia Britannica 1911
Wikipedia for Schools
Bach Organ Works
Audioletture
May 2024 M T W T F S S 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31