

Fibrose kystique
Contexte des écoles Wikipédia
Enfants SOS offrent un chargement complet de la sélection pour les écoles pour une utilisation sur les intranets des écoles. SOS Children travaille dans 45 pays africains; pouvez-vous aider un enfant en Afrique ?
| Fibrose kystique | |
|---|---|
| les ressources de classification et externes | |
| CIM 10 | E 84 |
| CIM 9 | 277,0 |
| OMIM | 219700 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| eMedicine | ped / 535 |
| MeSH | D003550 |
La fibrose kystique (aussi connu comme la mucoviscidose ou CF) est un autosomique récessif maladie génétique qui affecte le plus critique de la les poumons, et également la pancréas, foie, et intestin. Elle est caractérisée par le transport anormal de et chlorure de sodium à travers un épithélium, conduisant à, sécrétions épaisses et visqueuses.
Le nom de fibrose kystique renvoie au caractère cicatrices ( fibrose) et la formation de kystes dans le du pancréas, d'abord reconnus dans les années 1930. Difficulté à respirer est le symptôme le plus grave et résulte de fréquentes infections pulmonaires qui sont traités avec des antibiotiques et autres médicaments. Autre symptômes, y compris infections des sinus, faible croissance, et infertilité affecte d'autres parties du corps.
CF est causée par un mutation dans le gène de la protéine régulateur transmembranaire de la fibrose kystique conductance (CFTR). Cette protéine est nécessaire pour réglementer les composants de la sueur, sucs digestifs, et mucus. CFTR régule le mouvement de et chlorure de sodium des ions à travers les membranes epitheliales, telles que l'épithélium alvéolaire situés dans la les poumons. Bien que la plupart des gens sans FC ont deux copies de travail du gène CFTR, un seul est nécessaire pour prévenir la fibrose kystique en raison de la nature récessive de la maladie. CF développe lorsque ni gène fonctionne normalement (en raison de mutation) et a donc autosomique transmission récessive.
CF est plus fréquent chez les Caucasiens; Une personne sur 25 personnes d'origine européenne une porte allèle de la mucoviscidose L' Organisation mondiale de la Santé déclare que "Dans l'Union européenne, dans une nouveau-nés de 2000 à 3000 se trouve être affecté par CF". Personnes atteintes de fibrose kystique peuvent être diagnostiquées avant la naissance par les tests génétiques, ou par un test de la sueur dans la petite enfance. En fin de compte, La transplantation pulmonaire est souvent nécessaire que CF aggrave.
Signes et symptômes

Les signes et les symptômes de la fibrose kystique Hallmark sont dégustation salée la peau, une mauvaise croissance et le gain de poids mauvaise malgré une alimentation normale, l'accumulation de mucus épais et collant, infections pulmonaires fréquentes, et de la toux ou de l'essoufflement. Les mâles peuvent être infertiles en raison d' absence congénitale des canaux déférents. Les symptômes apparaissent souvent dans la petite enfance et l'enfance, tels que occlusion intestinale due à iléus méconial des nouveau-nés. Comme les enfants grandissent, ils doivent exercer à libérer le mucus dans les alvéoles. Ciliée cellules épithéliales chez le patient ont une protéine mutée qui conduit à la production de mucus anormalement visqueux. La faible croissance chez les enfants présente généralement comme une incapacité à prendre du poids ou de la hauteur au même rythme que leurs pairs et est parfois pas diagnostiquée avant enquête est ouverte pour une faible croissance. Les causes de l'échec de croissance sont multifactorielles et comprennent l'infection pulmonaire chronique, mauvaise absorption des nutriments à travers le tractus gastro-intestinal, et l'augmentation de la demande métabolique due à une maladie chronique.
Dans de rares cas, la fibrose kystique peut se manifester comme un trouble de la coagulation. Un allèle récessif deux est nécessaire pour la fibrose kystique à être apparente. Les jeunes enfants sont particulièrement sensibles à la vitamine K troubles de malabsorption parce que seule une très petite quantité de vitamine K traverse le placenta, laissant l'enfant avec de très faibles réserves. Parce que les facteurs II, VII, IX et X (facteurs de coagulation) sont la vitamine K-dépendant, de faibles niveaux de vitamine K peut entraîner des problèmes de coagulation. Par conséquent, lorsqu'un enfant présente des ecchymoses inexpliquées, une évaluation de la coagulation peut être justifié de déterminer se il existe une maladie sous-jacente.
Poumons et les sinus

= Vert Pseudomonas aeruginosa
Brown = Staphylococcus aureus
Bleu = Haemophilus influenzae
= Rouge Burkholderia cepacia
Les résultats de la maladie du poumon de colmatage des voies respiratoires due à l'accumulation de mucus, diminué clairance mucociliaire, et résultant l'inflammation. L'inflammation et l'infection cause des blessures et des changements structurels dans les poumons, conduisant à une variété de symptômes. Dans les premiers stades, toux incessante, copieux la production de flegme, et diminution de la capacité d'exercer sont communs. Plusieurs de ces symptômes se produisent lorsque les bactéries qui habitent normalement les mucus épais poussent hors de contrôle et provoquer une pneumonie. Dans les stades ultérieurs, les changements dans l'architecture du poumon, telles que la pathologie dans les voies aériennes principales ( bronchectasie), qu'exacerber difficultés à respirer. D'autres symptômes incluent des crachats de sang ( hémoptysie), haute pression sanguine dans les poumons ( l'hypertension pulmonaire), insuffisance cardiaque, des difficultés à obtenir suffisamment d' oxygène au corps ( hypoxie), et une insuffisance respiratoire nécessitant un soutien avec des masques respiratoires, tels que à deux niveaux machines à pression positive ou ventilateurs. Staphylococcus aureus , Haemophilus influenzae, et Pseudomonas aeruginosa sont les trois organismes les plus communs provoquant des infections pulmonaires chez les patients atteints de mucoviscidose. En outre à des infections bactériennes typiques, personnes atteintes de FK plus communément développer d'autres types de maladies pulmonaires. Parmi ceux-ci est aspergillose broncho-pulmonaire allergique, dans lequel la réponse du corps à la commune champignon Aspergillus fumigatus provoque aggravation des problèmes respiratoires. Une autre est l'infection par Complexe Mycobacterium avium (MAC), un groupe de bactéries liées à la tuberculose , qui peut causer beaucoup de dommages aux poumons et ne répond pas aux antibiotiques courants.
Le mucus dans le sinus de la face est la même épaisseur et peut également entraîner une obstruction des sinus, conduisant à l'infection. Cela peut causer des douleurs du visage, fièvre, écoulement nasal et des maux de tête . Personnes atteintes de FK peuvent développer prolifération du tissu nasal ( polypes nasaux) en raison de l'inflammation des infections chroniques du sinus. Polypes naso-sinusales récurrentes peuvent se produire dans autant que 10% à 25% des patients atteints de mucoviscidose. Ces polypes peuvent bloquer les voies nasales et d'accroître les difficultés de respiration.
Complications cardiorespiratoires sont la cause la plus fréquente de décès (~ 80%) chez les patients à la plupart des centres des FC aux États-Unis.
Gastrointestinal
Avant prénatal et dépistage néonatal, la fibrose kystique a été souvent diagnostiqué quand un nouveau-né n'a pas à passer les selles ( méconium). Méconium peut complètement bloquer la intestins et causer des maladies graves. Cette condition, appelée iléus méconial, se produit dans 5 à 10% des nouveau-nés atteints de mucoviscidose En outre, la saillie de interne (membranes rectales prolapsus rectal) est plus fréquent, survenant dans autant que 10% des enfants atteints de mucoviscidose, et elle est causée par l'augmentation du volume fécal, la malnutrition et augmentation de la pression intra-abdominale due à la toux.
Le mucus épais vu dans les poumons a une contrepartie dans les sécrétions épaissies de la du pancréas, un organe chargé de fournir sucs digestifs qui aident décomposer les aliments. Ces sécrétions bloquent la mouvement exocrine du enzymes digestives dans le duodénum et entraîner des dommages irréversibles au pancréas, souvent avec une inflammation douloureuse ( pancréatite). Le canaux pancréatiques sont totalement branchés dans les cas plus avancés, généralement observés chez les enfants plus âgés ou les adolescents. Cela provoque l'atrophie des glandes exocrines et fibrose progressive.
Le manque d'enzymes digestives conduit à l'absorption des nutriments avec leur excrétion dans les selles difficulté, un trouble connu sous le nom malabsorption. Malabsorption conduit à la malnutrition et une mauvaise croissance et de développement en raison de la perte de calories. Résultant hypoprotéinémie peut être suffisamment grave pour provoquer un oedème généralisé. Personnes atteintes de FK ont aussi du mal à absorber les vitamines liposolubles A, D , E et K .
En plus des problèmes de pancréas, les personnes atteintes de fibrose kystique expérience de plus brûlures d'estomac, une obstruction intestinale par invagination, et la constipation. Les personnes âgées atteintes de la mucoviscidose peuvent se développer syndrome d'obstruction intestinale distale lorsque les matières fécales épaissies provoquent une occlusion intestinale.
L'insuffisance pancréatique exocrine se produit dans la majorité (85% à 90%) des patients atteints de CF. Il est principalement associée à des mutations du gène CFTR «graves», où les deux allèles sont complètement non fonctionnel (par exemple AF508 / AF508). Il se produit dans 10% à 15% des patients avec une «grave» et une mutation du gène CFTR «douce» où il ya encore un peu d'activité CFTR, ou où il ya deux mutations du gène CFTR «douces». Dans ces cas moins graves, il existe toujours la fonction exocrine pancréatique suffisante pour que la supplémentation en enzyme est pas nécessaire. Il ne existe généralement pas d'autres complications gastro-intestinales dans phénotypes de pancréas-suffisante, et en général, ces personnes ont généralement une excellente croissance et le développement. Malgré cela, idiopathique pancréatite chronique peut se produire dans un sous-ensemble d'individus de pancréas-suffisante avec CF, et est associée à des douleurs abdominales récurrentes et les complications potentiellement mortelles.
Sécrétions épaissies peuvent également causer des troubles hépatiques chez les patients atteints de mucoviscidose Bile sécrétée par le foie pour aider à la digestion peut bloquer le voies biliaires, conduisant à des dommages au foie. Au fil du temps, cela peut conduire à des cicatrices et des nodules ( cirrhose). Le foie ne parvient pas à débarrasser le sang des toxines et ne fait pas importantes protéines , tels que les responsables de la coagulation du sang. Les maladies du foie est la troisième cause la plus fréquente de décès associé à FC
Endocrine

Le contient le pancréas îlots de Langerhans, qui sont responsables de fabriquer de l'insuline, une hormone qui aide à réguler le sang glucose . Dommages du pancréas peut conduire à la perte de l'îlot des cellules , conduisant à un type de diabète qui est unique à ceux qui ont la maladie. Cette diabète lié à la fibrose kystique (CFRD) part des caractéristiques qui peuvent être trouvés dans type 1 et diabétiques de type 2, et est l'une des principales complications non pulmonaire de FC La vitamine D est impliquée dans calcium et la réglementation phosphate. Mauvaise utilisation de la vitamine D dans l'alimentation en raison de malabsorption peut conduire à la maladie des os l'ostéoporose chez qui affaiblit les os sont plus sensibles à fractures. En outre, les personnes atteintes de mucoviscidose développent souvent Clubbing de leurs doigts et les orteils en raison des effets de la maladie chronique et faible teneur en oxygène dans les tissus.
Infertilité
Infertilité affecte les hommes et les femmes. Au moins 97% des hommes atteints de fibrose kystique sont infertiles, mais pas stérile et peut avoir des enfants avec des techniques de procréation assistée. La principale cause de l'infertilité chez les hommes atteints de fibrose kystique est absence congénitale des canaux déférents (qui relie normalement la testicules au canaux éjaculateurs de la pénis), mais potentiellement aussi par d'autres mécanismes tels que causer azoospermie, tératospermie et oligoasthenospermia. Beaucoup d'hommes ont trouvé à l'absence congénitale des canaux déférents lors de l'évaluation de l'infertilité ont une forme bénigne, non diagnostiquée auparavant de la mucoviscidose Certaines femmes ont des difficultés de fertilité dus à la malnutrition ou le mucus cervical épaissies. Dans les cas graves, perturbe de malnutrition ovulation et les causes aménorrhée.
Cause
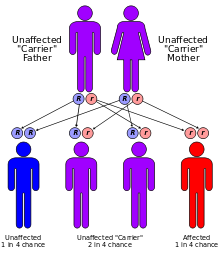
CF est causée par un mutation dans le gène régulateur transmembranaire de la fibrose kystique conductance (CFTR). La mutation la plus commune, AF508, est une délétion ( Δ signifie la suppression) de trois nucléotides qui se traduit par une perte de l'acide aminé la phénylalanine (F) à la position 508e sur la protéine. Ce comptes de mutation pour les deux tiers (66 à 70%) des cas des FC à travers le monde et 90% des cas dans le États-Unis ; Cependant, il existe plus de 1500 mutations d'autres qui peuvent produire CF. Bien que la plupart des gens ont deux copies de travail (allèles) du gène CFTR, un seul est nécessaire pour prévenir la fibrose kystique. CF développe lorsque ni l'allèle peut produire une protéine CFTR fonctionnelle. Ainsi, CF est considéré comme un maladie récessive autosomique.
Le Gène CFTR, se trouve à la q31.2 locus de chromosome 7, est 230000 paires de bases de long, et crée une protéine qui est 1480 acides aminés de long. Plus précisément l'emplacement se situe entre la paire de base 117 120 016 117 308 718 à sur le bras long du chromosome 7, la région 3, bande 1, sous-bande 2, représentée comme 7q31.2. Structurellement, la protéine CFTR est un type de gène connu comme un Gène ABC. Le produit de ce gène (CFTR) est un canal d'ions chlorure important dans la création sueur, sucs digestifs et mucus. Cette protéine possède deux Hydrolyser l'ATP domaines, ce qui permet à la protéine d'utiliser l'énergie sous forme d' ATP . Il contient également deux domaines comprenant 6 hélices alpha, qui permettent chacun la protéine de traverser la membrane cellulaire. Un réglementaire site de liaison de la protéine permet activation par phosphorylation, principalement par AMPc-dépendante protéine kinase. Le carboxy terminale de la protéine est fixée à la par un cytosquelette PDZ domaine interaction.
En outre, il existe des preuves croissantes que modificateurs génétiques outre CFTR modulent la fréquence et la gravité de la maladie. Un exemple est lectine mannane, qui est impliqué dans l'immunité innée en facilitant la phagocytose des micro-organismes. Polymorphismes dans un ou deux allèles de lectine mannane qui se traduisent par la baisse des taux circulants de la protéine sont associés à un risque trois fois plus élevé d'une maladie pulmonaire en phase terminale, ainsi que par un fardeau accru d'infections bactériennes chroniques.
Physiopathologie

Il existe plusieurs mutations dans le gène CFTR, et différentes mutations provoquent différents défauts de la protéine CFTR, ce qui provoque parfois une maladie plus ou moins grave. Ces défauts de protéines sont également des cibles pour les médicaments qui peuvent parfois restaurer leur fonction. AF508-CFTR, qui se produit dans> 90% des patients aux États-Unis, crée une protéine qui ne est pas plier normalement et est dégradée par la cellule. D'autres mutations conduisent à des protéines qui sont trop courts (tronqué), car la production est terminée prématurément. D'autres mutations produisent des protéines qui ne utilisent pas l'énergie normalement, ne permettent pas chlorure, iodure et thiocyanate de traverser la membrane appropriée, ou se dégradent à un rythme plus rapide que la normale. Des mutations peuvent également conduire à moins de copies de la protéine CFTR produite.
La protéine créée par ce gène est ancrée à la la membrane externe de cellules dans le les glandes sudoripares, les poumons, le pancréas et les glandes exocrines autres restant dans le corps. La protéine se étend sur cette membrane et agit comme un canal reliant la partie interne de la cellule ( cytoplasme) de la fluide environnant. Cette chaîne est principalement responsable de la commande du mouvement d'halogènes de l'intérieur vers l'extérieur de la cellule; cependant, dans les canaux sudoripares elle facilite le mouvement de chlorure de la sueur dans le cytoplasme. Lorsque la protéine CFTR ne fonctionne pas, le chlorure et le thiocyanate sont piégés à l'intérieur des cellules dans les voies aériennes et à l'extérieur de la peau. Puis hypothiocyanite, OSCN, ne peut être produite par le système de défense immunitaire. Parce que le chlorure est chargée négativement , ce qui crée une différence de potentiel électrique à l'intérieur et à l'extérieur de la cellule provoquant cations de traverser dans la cellule. Le sodium est le cation le plus commun dans l'espace extracellulaire et la combinaison de sodium et de chlorure crée le sel , qui est perdu en grande quantité dans la sueur des individus atteints de mucoviscidose Ce sel perdu constitue la base pour le test de la sueur.
La plupart des dommages dans les FC est en raison de l'obstruction des passages étroits des organes touchés avec des sécrétions épaisses. Ces blocages conduisent à un remodelage et d'infection dans les poumons, les dommages causés par les enzymes digestives accumulés dans le pancréas, le blocage de l'intestin par des matières fécales d'épaisseur, etc. Il existe plusieurs théories sur la façon dont les défauts de la protéine et la fonction cellulaire provoquent les effets cliniques. Une théorie est que le manque d'halogène et pseudo (principalement, chlorure, iodure et thiocyanate) de sortir par la protéine CFTR conduit à l'accumulation de mucus riche en éléments nutritifs plus visqueux dans les poumons qui permet aux bactéries de se cacher de l'organisme système immunitaire . Une autre théorie est que l'échec de la protéine CFTR conduit à une augmentation paradoxale de l'absorption de sodium et de chlorure, qui, en conduisant à une augmentation de la réabsorption de l'eau, crée le mucus déshydraté et épaisse. Une autre théorie est que le mouvement de chlorure anormale de la cellule conduit à une déshydratation du mucus, les sécrétions pancréatiques, sécrétions biliaires, etc.
Les infections chroniques
Les poumons des personnes atteintes de fibrose kystique sont colonisées et infectées par des bactéries à un âge précoce. Ces bactéries, qui se propagent souvent chez les individus atteints de mucoviscidose, se développent dans le mucus modifiée, qui se accumule dans les petites voies respiratoires des poumons. Ce mucus conduit à la formation de micro-environnements bactériens connus comme biofilms qui sont difficiles pour les cellules et les antibiotiques pour pénétrer immunitaires. Sécrétions visqueuses et les infections respiratoires persistants endommagent plusieurs reprises du poumon en remodelant progressivement les voies respiratoires, ce qui rend l'infection encore plus difficile à éradiquer.
Au fil du temps, les deux types de bactéries et de leurs caractéristiques individuelles changent chez les individus atteints de mucoviscidose Dans la phase initiale, les bactéries courantes telles que Staphylococcus aureus et Haemophilus influenzae coloniser et infecter les poumons. Finalement, Pseudomonas aeruginosa (et parfois Burkholderia cepacia) domine. En 18 ans, 80% des patients atteints de CF classique port P. aeruginosa, et 3,5% port B. cepacia. Une fois dans les poumons, ces bactéries se adaptent à l'environnement et à développer résistance aux antibiotiques couramment utilisés. Pseudomonas peut développer des caractéristiques spéciales qui permettent la formation de grandes colonies, appelées Pseudomonas "mucoïdes», qui sont rarement vus chez les personnes qui ne ont pas CF.
Une façon infection se propage est en passant entre différents individus atteints de mucoviscidose Dans le passé, les gens atteints de mucoviscidose souvent participé à été "FC" Camps de loisirs et autres rassemblements. Hôpitaux regroupés les patients atteints de mucoviscidose dans les zones communes et les équipements de routine (comme nébuliseurs) n'a pas été stérilisés entre les patients individuels. Cela a conduit à la transmission de souches de bactéries les plus dangereuses au sein des groupes de patients. En conséquence, les personnes atteintes de FK sont systématiquement isolés les uns des autres dans le cadre des soins de santé et les fournisseurs de soins de santé sont encouragés à porter des blouses et des gants lors de l'examen des patients atteints de mucoviscidose pour limiter la propagation de souches bactériennes virulentes.
Patients atteints de mucoviscidose peuvent aussi avoir leurs voies respiratoires chroniques colonisées par des champignons filamenteux (comme Aspergillus fumigatus, Apiospermum Scedosporium, Aspergillus terreus) et / ou des levures (telles que Candida albicans); autres champignons filamenteux moins souvent isolés comprennent Aspergillus flavus et Aspergillus nidulans (survenir de façon transitoire dans les sécrétions respiratoires FC), et Dermatitidis et Exophiala prolificans de Scedosporium (voies aériennes des colonisateurs chroniques); des champignons filamenteux tels que Pénicillium emersonii et Fusispora Acrophialophora sont rencontrées chez les patients presque exclusivement dans le cadre de la mucoviscidose Clairance mucociliaire défectueux caractériser CF est associée à des troubles immunologiques locaux. En outre, le traitement prolongé avec des antibiotiques et l'utilisation de traitements de corticostéroïdes peuvent également faciliter la croissance fongique. Bien que la pertinence clinique de la colonisation des voies aériennes fongique est encore un sujet de débat, des champignons filamenteux peuvent contribuer à la réponse inflammatoire locale, et donc à la détérioration progressive de la fonction pulmonaire, comme cela arrive souvent avec aspergillose broncho-pulmonaire allergique (ABPA) - la maladie fongique la plus courante dans le cadre de la mucoviscidose, impliquant une réponse immunitaire Th2 à entraînée Aspergillus.
Le diagnostic et le suivi
La fibrose kystique peut être diagnostiqué par de nombreuses méthodes différentes, y compris dépistage néonatal, transpirer tests, et tests génétiques. En 2006 aux États-Unis, 10 pour cent des cas sont diagnostiqués peu après la naissance dans le cadre de programmes de dépistage néonatal. L'écran du nouveau-né mesure initialement pour la concentration sanguine élevée de trypsinogène immunoréactive. Les nourrissons atteints d'un écran nouveau-né anormale besoin d'un test de la sueur pour confirmer le diagnostic de FC. Dans de nombreux cas, un parent rend le diagnostic parce que le nourrisson goût salé. Trypsinogène niveaux peuvent être augmentés chez les personnes qui ont une copie mutée unique du gène CFTR (transporteurs) ou, dans de rares cas, chez les personnes ayant deux copies normales du gène CFTR. En raison de ces faux positifs, dépistage de la FK chez les nouveaux nés peuvent être controversées. La plupart des États et pays ne permet pas de dépister FC systématiquement à la naissance. Par conséquent, la plupart des individus sont diagnostiqués après que les symptômes (par exemple, les maladies et les GI manifestations sinopulmonaires) demandera une évaluation de la fibrose kystique. La forme la plus couramment utilisée de test est le test de la sueur. Sweat-test consiste en l'application d'un médicament qui stimule la transpiration ( pilocarpine). Pour administrer le médicament à travers la peau, iontophorèse est utilisé pour, par lequel une électrode est placée sur le médicament appliquée et un courant électrique est passé à une électrode distincte sur la peau. La sueur résultant est ensuite recueilli sur un papier filtre ou dans un tube capillaire et analysé pour déterminer des quantités anormales de sodium et chlorure. Personnes atteintes de FK ont augmenté quantités de sodium et de chlorure dans la sueur. En revanche, les personnes atteintes de FK ont moins thiocyanate et hypothiocyanite dans la salive et le mucus (Banfi et al.). CF peut également être diagnostiquée par l'identification de mutations dans le gène CFTR.
Personnes atteintes de FK peuvent être énumérés dans un Registre de la maladie qui permet aux chercheurs et aux médecins de suivre les résultats de santé et identifier les candidats pour essais cliniques.
Prénatal
Les couples qui sont enceintes ou qui prévoient une grossesse peuvent se sont testés pour les mutations du gène CFTR pour déterminer le risque que leur enfant sera né avec la fibrose kystique. Le test est généralement effectué d'abord sur un ou deux parents et, si le risque de CF est élevé, les tests sur le foetus est effectuée. Le Collège américain des obstétriciens et gynécologues (ACOG) recommande de tester pour les couples qui ont des antécédents personnels ou familiaux de CF, et ils recommandent que le test de support sera offert à tous les couples de race blanche et être mis à la disposition des couples d'autres origines ethniques.
Parce que le développement de la mucoviscidose chez le fœtus nécessite chaque parent de transmettre une copie mutée du gène CFTR et parce que les tests CF est cher, les tests sont souvent effectués initialement sur un parent. Si le test montre que parent est porteur d'une mutation du gène CFTR, l'autre parent est testé pour calculer le risque que leurs enfants auront FC FC peut entraîner à partir de plus d'un millier de différentes mutations, et à partir de 2006, il ne est pas possible de tester pour chacun. Test analyse le sang pour les mutations les plus courantes telles que AF508-plus commercialement tests disponibles recherchent ou moins 32 mutations différentes. Si une famille a connu une mutation rare, le dépistage spécifique pour que la mutation peut être effectuée. Parce que pas toutes les mutations connues se trouvent sur les tests actuels, un écran négatif ne garantit pas que l'enfant ne aura pas CF.
Pendant la grossesse, le test peut être effectué sur la placenta ( prélèvement de villosités choriales) ou le fluide entourant le fœtus ( amniocentèse). Cependant, prélèvement de villosités choriales a un risque de mort fœtale de 1 à 100 et l'amniocentèse de 1 sur 200; une étude récente a indiqué cela peut être beaucoup plus faible, environ 1 sur 1600.
Economiquement, pour les couples de porteuses de fibrose kystique, lorsque l'on compare le diagnostic génétique préimplantatoire (DPI) à une conception naturelle (NC), suivie par le dépistage prénatal et l'avortement des grossesses en cause, le DPI fournit des avantages économiques nets jusqu'à un âge maternel d'environ 40 ans, après quoi NC, le dépistage prénatal et l'avortement a avantage économique supérieur.
Gestion
Bien qu'il ne existe aucun remède pour la mucoviscidose, il existe plusieurs méthodes de traitement. La gestion de la fibrose kystique se est considérablement améliorée au cours des 70 dernières années. Pendant que les enfants nés avec la fibrose kystique il ya 70 ans auraient été susceptibles de vivre au-delà de leur première année, les bébés d'aujourd'hui sont susceptibles de bien vivre à l'âge adulte. Les progrès récents dans le traitement de la fibrose kystique ont signifié qu'une personne atteinte de fibrose kystique peut vivre une vie plus pleine moins encombrés par leur état. Les pierres angulaires de la gestion proactive de traitement sont des voies respiratoires infection, et l'encouragement d'une bonne nutrition et une vie active. Gestion de la fibrose kystique continue tout au long de la vie d'un patient, et vise à maximiser la fonction des organes, et donc la qualité de la vie. Au mieux, les traitements actuels retarder le déclin de la fonction d'organe. En raison de la grande variation dans la maladie un traitement des symptômes se produit généralement dans les centres multidisciplinaires de spécialistes, et est adapté à l'individu. Cibles pour la thérapie sont les les poumons, tractus gastro-intestinal (y compris les suppléments d'enzymes pancréatiques), le organes de reproduction (y compris la technologie de reproduction assistée (ART)) et un soutien psychologique.
L'aspect le plus uniforme de la thérapie de la mucoviscidose et le traitement est de limiter les dégâts causés par des poumons mucus épais et de l'infection, avec l'objectif de maintenir qualité de vie. Intraveineuse, inhalées, et des antibiotiques oraux sont utilisés pour traiter les infections chroniques et aiguës. Les dispositifs mécaniques et des médicaments par inhalation sont utilisés pour modifier et effacer le mucus épaissi. Ces traitements, bien qu'ils soient efficaces, peuvent être extrêmement fastidieux pour le patient. L'une des batailles les plus importantes qui FC patients confrontés est de trouver le temps pour se conformer avec les traitements prescrits tout en équilibrant une vie normale.
En outre, des thérapies telles que transplantation et la thérapie génique pour but de guérir certains des effets de la fibrose kystique. La thérapie génique vise à introduire CFTR normal de voies aériennes. Théoriquement ce processus devrait être simple que la voie aérienne est facilement accessible et il ya seulement un défaut d'un seul gène à corriger. Il existe deux mécanismes d'introduction du gène CFTR impliqués, la première utilisation d'un vecteur viral (adénovirus, virus adéno-associé ou rétro virus) et d'autre part l'utilisation de liposome. Cependant, il ya certains problèmes associés à ces procédés impliquant efficacité (liposomes de protéines insuffisante) et la livraison (le virus provoque une réponse immunitaire).
Antibiotiques
De nombreux patients atteints de mucoviscidose sont sur un ou plusieurs antibiotiques en tout temps, même lorsque la santé, à prophylactique supprimer l'infection. Les antibiotiques sont absolument nécessaires chaque fois on soupçonne une pneumonie ou il ya eu une baisse notable de la fonction pulmonaire, et sont généralement choisi sur la base des résultats d'une analyse des expectorations et la réponse passé du patient. Cette thérapie prolongée nécessite souvent une hospitalisation et l'insertion d'un plus permanent IV tel qu'un cathéter central inséré par voie périphérique (PICC line) ou Port-a-Cath. Le traitement par inhalation avec des antibiotiques tels que la tobramycine, colistine, et aztréonam est souvent donnée pendant des mois à la fois pour améliorer la fonction pulmonaire en empêchant la croissance des bactéries colonisés. Les antibiotiques oraux tels que la ciprofloxacine ou azithromycine sont donnés pour aider à prévenir l'infection ou à contrôler l'infection en cours. Le aminosides (par exemple la tobramycine) utilisés peuvent causer la perte, des dommages auditifs à la système d'équilibre dans le oreille interne ou des problèmes rénaux avec l'utilisation à long terme. Pour éviter ces effets secondaires, la quantité d'antibiotiques dans le sang sont régulièrement mesurés et ajustés en conséquence.
D'autres traitements pour les maladies pulmonaires
Plusieurs techniques mécaniques sont utilisées pour déloger crachats et d'encourager son expectoration. Dans le milieu hospitalier, la kinésithérapie respiratoire (CPT) est utilisé; un inhalothérapeute percute la poitrine d'un individu avec ses mains plusieurs fois par jour, pour desserrer les sécrétions. Dispositifs qui recréent cette thérapie de percussion comprennent la ThAIRapy Vest et la ventilateur percussion intrapulmonaire (VPI). De nouvelles méthodes telles que Cuirasse de ventilation biphasique, et le mode de dégagement associée disponible dans de tels dispositifs, à intégrer une phase d'aide à la toux, ainsi que d'une phase de vibration pour déloger les sécrétions. Ce sont portables et adapté pour un usage domestique.
Médicaments en aérosol qui aident desserrer les sécrétions comprennent dornase alfa et hypertonique une solution saline. Est une dornase humaine recombinante désoxyribonucléase, qui se décompose dans l'ADN crachat, ce qui diminue son viscosité. Denufosol est un médicament expérimental qui ouvre un canal de chlorure de remplacement, aidant à liquéfier le mucus.
Comme une maladie pulmonaire se aggrave, le soutien respiratoire mécanique peut être nécessaire. Les personnes atteintes de fibrose kystique peuvent avoir besoin de porter des masques spéciaux de nuit qui aident à l'air de poussée dans leurs poumons. Ces machines, appelées à deux niveaux de pression positive (BiPAP) ventilateurs, aident à prévenir les faibles niveaux d'oxygène dans le sang pendant le sommeil. BiPAP peut également être utilisé pendant la thérapie physique pour améliorer la clairance de crachats. Au cours de maladie grave, un tube peut être placé dans la gorge (une procédure connue comme une trachéotomie) pour permettre la respiration soutenue par un ventilateur.
Pour les enfants vivant avec le FC, des études préliminaires montrent la thérapie de massage pédiatrique peut améliorer les patients et leur qualité de vie des familles, mais des études plus rigoureuses doivent être effectuées.
Transplantation
La transplantation pulmonaire devient souvent nécessaire pour les personnes atteintes de fibrose kystique que la fonction pulmonaire et tolérance à l'effort diminue. Bien que la transplantation pulmonaire simple est possible dans d'autres maladies, personnes atteintes de FK doivent avoir deux poumons remplacés parce que le poumon reste pourrait contenir des bactéries qui pourraient infecter les poumons transplantés. Une transplantation du pancréas ou du foie peut être effectuée en même temps afin de soulager une maladie du foie et / ou le diabète. La transplantation pulmonaire est envisagée lorsque la fonction pulmonaire diminue au point où l'aide de dispositifs mécaniques est nécessaire ou la survie du patient est menacée.
Autres aspects

Les nouveau-nés présentant une obstruction intestinale nécessitent généralement une intervention chirurgicale, alors que les adultes avec syndrome d'obstruction intestinale distale faire généralement pas. Traitement de l'insuffisance pancréatique par remplacement d'enzymes digestives manquantes permet le duodénum à absorber correctement les nutriments et vitamines qui autrement seraient perdus dans le feces.So présent, aucune recherche à grande échelle impliquant l'incidence de l'athérosclérose et de maladie coronarienne chez les adultes atteints de fibrose kystique a été menée. Cela est probablement dû au fait que la grande majorité des personnes atteintes de fibrose kystique ne vivent pas assez longtemps pour développer une athérosclérose cliniquement significative ou une maladie coronarienne.
Le diabète est la complication la non-pulmonaire la plus courante de la mucoviscidose Il associe caractéristiques de type 1 et le diabète de type 2, et est reconnu comme une entité distincte, diabète lié à la fibrose kystique (CFRD). Bien orale médicaments anti-diabétiques sont parfois utilisés, le seul traitement recommandé est l'utilisation de l'insuline ou des injections d'un pompe à insuline, et, contrairement à de type 1 et le diabète 2, les restrictions alimentaires ne sont pas recommandés.
Développement de l'ostéoporose peut être prévenue par un apport accru en vitamine D et de calcium , et peut être traitée par bisphosphonates, bien effets indésirables peuvent être un problème. Une mauvaise croissance peut être évitée par l'insertion d'un sonde d'alimentation pour augmenter calories par une alimentation supplémentaire ou par administration d'injection l'hormone de croissance.
Infections des sinus sont traités par des cours prolongées d'antibiotiques. Le développement de polypes nasaux ou d'autres changements chroniques dans les voies nasales peut considérablement limiter le flux d'air par le nez, et au fil du temps réduire le sens de l'odorat du patient. la chirurgie des sinus est souvent utilisé pour soulager l'obstruction nasale et de limiter les nouvelles infections. Stéroïdes nasaux tels que fluticasone sont utilisés pour diminuer l'inflammation nasale.
L'infertilité féminine peut être surmontée par la technologie de reproduction assistée, en particulier Les techniques de transfert d'embryon. La stérilité masculine due à l'absence de canal déférent peuvent être surmontés avec extraction testiculaire de sperme (TEST), la collecte de spermatozoïdes directement des testicules. Si l'échantillon prélevé contient trop peu de spermatozoïdes susceptibles d'avoir un spontanée fécondation, injection intracytoplasmique de spermatozoïdes peut être effectuée. Reproduction tiers est aussi une possibilité pour les femmes atteintes de mucoviscidose
Pronostic
Le pronostic de la mucoviscidose est améliorée grâce à un diagnostic plus précoce grâce au dépistage, un meilleur traitement et l'accès aux soins de santé. En 1959, l'âge médian de survie des enfants atteints de mucoviscidose aux États-Unis était de six mois. En 2008, la survie moyenne 37,4 années. Au Canada, la médiane de survie est passé de 24 années en 1982 à 47,7 en 2007.
Parmi ceux qui ont la fibrose kystique qui sont plus de 18 ans à partir de 2009; 92% avaient terminé l'école secondaire, 67% avaient au moins fait des études collégiales, 15% ont été désactivé et 9% étaient sans emploi, 56% étaient célibataires et 39% ont été mariés ou vivant avec un partenaire. En Russie l'âge médian de l'ensemble des patients est de 25, qui est causée par l'absence ou le coût élevé des médicaments et du fait que la transplantation pulmonaire est pas effectuée.
Qualité de vie
Les maladies chroniques peuvent être très difficiles à gérer. La fibrose kystique (FK) est une maladie chronique qui affecte les «voies digestives et respiratoires résultant de la malnutrition généralisée et les infections respiratoires chroniques". Les sécrétions épaisses bouchent les voies respiratoires, qui provoquent souvent des inflammations et les infections pulmonaires sévères. Par conséquent, le mucus rend difficile de respirer. Si elle est compromise, elle affecte la qualité de la vie de quelqu'un avec CF, et leur capacité à compléter des tâches telles que les tâches quotidiennes. Il est important pour les patients atteints de mucoviscidose à comprendre la relation néfaste que les maladies chroniques mettent sur la qualité de vie. Selon Schmitz et Goldbeck (2006), le fait que la fibrose kystique augmente de manière significative le stress émotionnel à la fois sur l'individu et la famille », et la routine quotidienne de traitement nécessaire de temps peut avoir des effets négatifs sur la qualité de vie (QOL)". Cependant, Havermans et ses collègues (2006) ont montré que les jeunes patients ambulatoires atteints de mucoviscidose qui ont participé à la CFQ-R (Cystic Fibrosis Questionnaire-Revised) "évaluant certains domaines de qualité de vie plus élevé que les leurs parents". Par conséquent, les patients ambulatoires atteints de mucoviscidose ont une vision plus positive d'eux-mêmes. En outre, il existe de nombreuses façons d'améliorer la qualité de vie chez les patients des FC. L'exercice est promu à augmenter la fonction pulmonaire. Le fait d'intégrer un régime d'exercice dans la routine quotidienne du patient CF peut améliorer considérablement la qualité de vie. Il n'y a pas de remède définitif pour la fibrose kystique. Cependant, il ya divers médicaments utilisés tels que, mucolytiques, les bronchodilatateurs, les stéroïdes et les antibiotiques qui ont pour but de mucus desserrage, l'expansion des voies respiratoires, diminution de l'inflammation et de lutte contre les infections pulmonaires.
Épidémiologie
| Mutation | Fréquence partout dans le monde |
|---|---|
| AF508 | 66% -70% |
| G542X | 2,4% |
| G551D | 1,6% |
| N1303K | 1,3% |
| W1282X | 1,2% |
| Tous les autres | 27,5% |
La fibrose kystique est la autosomique mortelle maladie récessive la plus fréquente chez les personnes de race blanche patrimoine. Aux États-Unis, environ 30.000 personnes ont FC; la plupart sont diagnostiqués par six mois d'âge. Dans le Canada , il ya environ 3500 personnes atteintes de mucoviscidose Environ 1 dans 25 personnes d'origine européenne, et un dans 30 des Américains de race blanche, est porteuse d'une mutation de la fibrose kystique. Bien que CF est moins fréquente chez ces groupes, d'environ 1 sur 46 Hispaniques, 1 à 65 Africains et 1 dans 90 Asiatiques portent au moins un gène CFTR anormale. L'Irlande a plus forte incidence dans le monde de la fibrose kystique, à 1: 1353.
Bien que techniquement une maladie rare, la fibrose kystique est classée comme l'une des maladies génétiques les d'abréger la vie les plus répandues. Elle est plus fréquente parmi les nations dans le monde occidental. Une exception est la Finlande , où un seul de 80 personnes porteurs d'une mutation FC. Aux États-Unis, 1 en 4000 enfants naissent avec CF. En 1997, environ 1 sur 3300 caucasien enfants aux États-Unis est né avec la fibrose kystique. En revanche, seulement 1 sur 15 000 enfants afro-américains souffrent de la fibrose kystique, et Américains d'origine asiatique, le taux était encore plus bas à 1 dans 32.000.
La mucoviscidose est diagnostiquée chez les hommes et les femmes de manière égale. Pour des raisons qui demeurent obscures, les données ont montré que les hommes ont tendance à avoir une plus longue espérance de vie que les femmes, mais des études récentes suggèrent que ce fossé entre les sexes ne peut plus exister peut-être en raison de l'amélioration des installations de soins de santé, alors qu'une étude récente de l'Irlande a identifié un lien entre l'œstrogène et de moins bons résultats dans les FC
La distribution des allèles CF varie selon les populations. La fréquence des transporteurs AF508 a été estimé à 1: 200 dans le nord de la Suède, 1: 143 dans les Lituaniens, et 01h38 au Danemark. Aucun transporteurs AF508 ont été trouvés parmi 171 Finlandais et 151 Saami personnes. AF508 ne se produit en Finlande, mais il est un allèle minoritaire il. La fibrose kystique est connu pour se produire en seulement 20 familles (Généalogique) en Finlande.
Hypothèses sur la prévalence
Le AF508 mutation est estimée à jusqu'à l'âge de 52000 années. De nombreuses hypothèses ont été avancées pour expliquer pourquoi une telle mutation létale a persisté et se propager dans la population humaine. Autres maladies autosomiques récessives commun tels que la drépanocytose ont été trouvés de protéger les transporteurs d'autres maladies, un concept connu sous le nom hétérozygote avantage. Résistance à la suivante ont tous été proposés comme sources possibles d'avantage hétérozygote:
- Choléra: Avec la découverte que la toxine du choléra nécessite protéines CFTR hôte normales pour fonctionner correctement, il a émis l'hypothèse que les porteurs de gènes CFTR mutantes bénéficié de la résistance au choléra et d'autres causes de la diarrhée. D'autres études ont pas confirmé cette hypothèse.
- Typhoïde: Normale protéines CFTR sont également essentiels pour l'entrée de Salmonella Typhi dans les cellules, ce qui suggère que les porteurs de gènes CFTR mutantes pourraient être résistant à la fièvre typhoïde . Aucune in vivo étude n'a encore confirmé. Dans les deux cas, le faible niveau de la fibrose kystique en dehors de l'Europe, dans des endroits où la fois le choléra et la fièvre typhoïde sont endémiques, est pas immédiatement explicable.
- Diarrhée: Il a également émis l'hypothèse que la prévalence de la mucoviscidose en Europe pourrait être relié avec le développement de domestication du bétail. Dans cette hypothèse, porteurs d'un chromosome unique CFTR mutant avaient une certaine protection contre la diarrhée provoquée par l'intolérance au lactose, avant l'apparition des mutations qui ont créé la tolérance au lactose.
- Tuberculose: Une autre explication possible est que les porteurs du gène pourraient avoir une certaine résistance à la tuberculose.
Histoire

Il est supposé que CF est apparu vers 3000 av raison de la migration des peuples, mutations génétiques, et de nouvelles conditions de nourriture. Bien que l'ensemble du spectre clinique de la CF n'a pas été reconnu jusqu'à ce que les années 1930, certains aspects de CF ont été identifiés beaucoup plus tôt. En effet, la littérature de l'Allemagne et de la Suisse au 18e siècle averti Wehe dem Kind, das beim Kuß auf die Stirn de la schmekt, er ist verhext und muss de Sterbe chauve ou «Malheur à l'enfant qui goût salé à partir d'un baiser sur le front, car il est maudit et bientôt doit mourir, "reconnaissant le lien entre la perte de sel dans les FC et la maladie.
Au 19ème siècle, Carl von Rokitansky décrit un cas de mort fœtale avec péritonite méconiale, une complication de méconium iléus associée à la fibrose kystique. Iléus méconial a été décrite pour la première en 1905 par Karl Landsteiner. En 1936, Guido Fanconi a publié un document décrivant un lien entre la maladie cœliaque, la fibrose kystique du pancréas et bronchectasies.
En 1938, Dorothy Hansine Andersen a publié un article, "la fibrose kystique du pancréas et de sa relation à la maladie coeliaque: une étude clinique et pathologique," dans le American Journal of maladies de l'enfance . Elle fut le premier à décrire la fibrose kystique caractéristique du pancréas et à corréler avec le poumon et la maladie intestinale importante dans CF. Elle a également émis l'hypothèse que la première CF est une maladie récessive et le premier utilisé le remplacement d'enzymes pancréatiques pour traiter les enfants atteints. En 1952, Paul di Sant 'Agnese découvert des anomalies dans sueur les électrolytes; une test de la sueur a été développée et améliorée au cours de la prochaine décennie.
Le premier lien entre les FC et un autre marqueur (Paroxonase) a été trouvé en 1985, indiquant que existe un seul locus pour CF Hans Eiberg. En 1988, la première mutation de la mucoviscidose, AF508 a été découvert par Francis Collins, Lap-Chee Tsui et John R. Riordan le septième chromosome. Des recherches ultérieures ont trouvé plus de 1000 mutations différentes qui causent CF.
Parce que des mutations dans le gène CFTR sont généralement petits, de techniques génétiques classiques avaient été incapables de localiser avec précision le gène muté. En utilisant des marqueurs de protéines, des études gène liaison étaient en mesure de cartographier la mutation sur le chromosome 7. Chromosome-marche et techniques -jumping ont ensuite été utilisées pour identifier et séquence du gène. En 1989 Lap-Chee Tsui a dirigé une équipe de chercheurs de l' Hospital for Sick Children de Toronto qui a découvert le gène responsable de la FK La fibrose kystique est la première maladie génétique élucidée strictement le processus de génétique inverse.
Recherche
La thérapie génique
La thérapie génique a été explorée comme un remède potentiel de la fibrose kystique. Idéalement, la thérapie génique place une copie normale du gène CFTR dans les cellules affectées. Le transfert du gène CFTR dans les cellules normales de l'épithélium affectées aurait pour résultat la production de la protéine CFTR fonctionnel dans les cellules cibles, sans réactions indésirables ou une réponse inflammatoire. Des études ont montré que pour prévenir les manifestations pulmonaires de la fibrose kystique, seulement 5 à 10% de la quantité normale de CFTR l'expression du gène est nécessaire. Plusieurs approches ont été testées pour le transfert de gènes, tels que des liposomes et des vecteurs viraux dans des modèles animaux et des essais cliniques. Cependant, les deux procédés se sont avérés être des options de traitement relativement inefficaces. La raison principale est que très peu de cellules relever le vecteur et exprimer le gène, de sorte que le traitement a peu d'effet. En outre, des problèmes ont été notés dans l'ADNc recombinaison, de telle sorte que le gène introduit par le traitement est rendu inutilisable. Avec l'aide de la Cystic Fibrosis Trust, qui a une ligue de thérapeutes de gènes hautement professionnels, à la fois somatique et vecteur viral adéno-associé ont fait des progrès. Le Adenoviridae, ou plus communément connu comme le virus du rhume, est génétiquement modifié, permettant le gène CFTR à entrer dans les cellules pulmonaires.
Les petites molécules
Un nombre de de petites molécules qui visent à compenser différentes mutations du gène CFTR sont en cours de développement. Une approche consiste à développer des médicaments qui obtiennent le ribosome pour surmonter le codon d'arrêt et de synthétiser une protéine CFTR pleine longueur. Environ 10% de la mucoviscidose résultent d'un codon stop prématuré dans l'ADN, ce qui conduit à la terminaison précoce de la synthèse des protéines et des protéines tronquées. Ces médicaments ciblent mutations non-sens, tels que G542X, qui consiste en l'acide aminé glycine en position 542 est remplacé par un codon d'arrêt. Les antibiotiques aminoglycosides interfèrent avec la synthèse d'ADN et de correction d'erreur. Dans certains cas, ils peuvent causer la cellule de surmonter le codon d'arrêt, insérez un acide aminé aléatoire, et d'exprimer une protéine pleine longueur. Le aminosides gentamicine a été utilisé pour traiter les cellules pulmonaires de patients atteints de mucoviscidose dans le laboratoire pour induire les cellules à croître protéines de pleine longueur. Un autre médicament ciblant mutations non-sens est ataluren, qui est en pleine phase III des essais cliniques comme d'Octobre 2011.
Ivacaftor (Kalydeco), approuvé pour une utilisation par la FDA aux États-Unis en Janvier 2012, vise la mutation G551D (glycine en position 551 est substitué par unacide aspartique).lumacaftor vise àF508del (phénylalanine en position 508 est manquant).