
Fibrose cística
Fundo para as escolas Wikipédia
Crianças SOS oferecem um download completo desta seleção para as escolas para uso em escolas intranets. SOS Children trabalha em 45 países africanos; você pode ajudar uma criança em África ?
| Fibrose cística | |
|---|---|
| Classificação e recursos externos | |
| CID- 10 | E 84 |
| CID- 9 | 277,0 |
| OMIM | 219700 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| Medcenter | ped / 535 |
| MeSH | D003550 |
A fibrose cística (também conhecido como CF ou mucoviscidose) é um autossômica recessivo doença genética que afeta mais gravemente a pulmões, e também o pâncreas, fígado, e intestino. É caracterizada por transporte anormal de e cloreto de sódio através de uma epitélio, levando a espessura, secreções viscosas.
O nome refere-se a fibrose cística a característica cicatriz ( fibrose) e formação de cistos dentro do pâncreas, reconhecida pela primeira vez na década de 1930. Dificuldade para respirar é o sintoma mais grave e resulta de frequentes infecções pulmonares que são tratados com antibióticos e outros medicamentos. Outro sintomas, incluindo sinusites, crescimento pobre, e infertilidade afectam outras partes do corpo.
FC é causada por um mutação no gene para a proteína transmembrana da fibrose cística reguladora da condutância (CFTR). Esta proteína é necessária para regular os componentes do suor, fluidos digestivos, e muco. CFTR regula a circulação de e cloreto de sódio iões através das membranas epiteliais, tais como os epitélios alveolares localizados no pulmões. Embora a maioria das pessoas sem CF têm duas cópias de trabalho do gene CFTR, apenas uma é necessária para prevenir a fibrose cística devido à natureza recessiva da doença. CF desenvolve quando nem gene funciona normalmente (como resultado de uma mutação) e, portanto, tem autossômica herança recessiva.
CF é mais comum entre Caucasianos; uma em 25 pessoas de ascendência europeia carrega uma alelo para CF. A Organização Mundial da Saúde afirma que "Na União Europeia, 1 em 2000-3000 recém-nascidos é encontrado para ser afetada por CF". Os indivíduos com fibrose cística pode ser diagnosticada antes do nascimento por testes genéticos, ou por um teste de suor na primeira infância. Em última análise, transplante pulmonar é muitas vezes necessário que se agrava CF..
Os sinais e sintomas
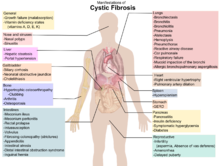
Os sinais e sintomas da fibrose cística da indicação são degustação salgado pele, problemas de crescimento e ganho de peso pobre, apesar de uma ingestão normal de alimentos, acúmulo de espessura, muco pegajoso, infecções respiratórias freqüentes e tosse ou falta de ar. Os machos podem ser inférteis devido à ausência congênita dos vasos deferentes. Os sintomas geralmente aparecem na infância, tais como obstrução intestinal devido a íleo meconial em recém-nascidos. À medida que as crianças crescem, elas devem exercer para liberar o muco nos alvéolos. Ciliado As células epiteliais em que o paciente tem uma proteína mutada que leva à produção de muco viscoso de forma anormal. O fraco crescimento em crianças apresenta tipicamente como uma incapacidade de ganhar peso ou a altura à mesma taxa que seus pares e é ocasionalmente não diagnosticada até investigação é iniciada para o crescimento pobre. As causas da falha do crescimento são multifatoriais e incluem infecção pulmonar crônica, má absorção de nutrientes pelo trato gastrointestinal, e aumento da demanda metabólica devido a doença crônica.
Em casos raros, a fibrose cística pode manifestar-se como um distúrbio de coagulação. Um alelo recessivo duplo é necessário para fibrose cística ser aparente. As crianças pequenas são especialmente sensíveis à vitamina K distúrbios de má absorção, porque apenas uma pequena quantidade de vitamina K atravessa a placenta, deixando a criança com reservas muito baixas. Como factores II, VII, IX e X (factores de coagulação) são a vitamina K-dependente, baixos níveis de vitamina K pode resultar em problemas de coagulação. Consequentemente, quando a criança apresentar hematomas inexplicável, uma avaliação de coagulação podem ser necessários para determinar se existe uma doença subjacente.
Pulmões e seios
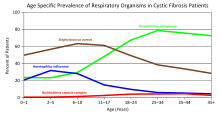
= Verde Pseudomonas aeruginosa
Brown = Staphylococcus aureus
= Azul Haemophilus influenzae
= Vermelho Complexo Burkholderia cepacia
Resultados da doença pulmonar de obstrução das vias aéreas devido ao acúmulo de muco, diminuição transporte mucociliar, e resultando inflamação. A inflamação causa a infecção e lesão e mudanças estruturais para os pulmões, que conduzem a uma variedade de sintomas. Nos estágios iniciais, incessante tosse, copioso produção de catarro, e diminuição da capacidade de exercício são comuns. Muitos desses sintomas ocorrem quando as bactérias que habitam normalmente o muco espesso crescer fora de controle e causar pneumonia. Em fases posteriores, mudanças na arquitetura do pulmão, como a patologia nas grandes vias aéreas ( bronquiectasia), agravar ainda mais dificuldades em respirar. Outros sintomas incluem tosse com sangue ( hemoptise), alta pressão arterial nos pulmões ( hipertensão pulmonar), insuficiência cardíaca, dificuldades recebendo o suficiente de oxigênio para o corpo ( hipoxia) e insuficiência respiratória necessitando de apoio com máscaras de respiração, tais como máquinas de dois níveis de pressão positiva nas vias aéreas ou ventiladores. Staphylococcus aureus , Haemophilus influenzae, e Pseudomonas aeruginosa são os três organismos mais comuns que causam infecções pulmonares em pacientes com FC. Para além típico de infecções bacterianas, as pessoas com CF mais comumente desenvolvem outros tipos de doença pulmonar. Entre elas está aspergilose broncopulmonar alérgica, em que a resposta do organismo à comum fungo Aspergillus fumigatus provoca o agravamento de problemas respiratórios. Outra é a infecção com Complexo Mycobacterium avium (MAC), um grupo de bactérias relacionadas à tuberculose , que pode causar uma série de danos pulmonares e não responde aos antibióticos comuns.
Muco no seios paranasais é igualmente de espessura e pode também causar o bloqueio das passagens dos seios nasais, levando à infecção. Isso pode causar dor facial, febre, drenagem nasal e dores de cabeça . Indivíduos com FC podem desenvolver overgrowth do tecido nasal ( pólipos nasais), devido à inflamação de infecções crônicas do seio. Pólipos sinonasais recorrentes podem ocorrer em até 10% a 25% de pacientes com FC. Estes pólipos podem bloquear as passagens nasais e aumentar a dificuldades respiratórias.
Complicações cardiorrespiratórias são a causa mais comum de morte (~ 80%) em pacientes na maioria dos centros de FC nos Estados Unidos.
Gastrointestinal
Antes do pré-natal e triagem neonatal, fibrose cística, foi frequentemente diagnosticada quando um recém-nascido não conseguiu passar fezes ( mecônio). Mecônio pode bloquear completamente a intestinos e causar doença grave. Esta condição, chamada íleo meconial, ocorre em 5-10% dos recém-nascidos com CF. Além disso, a saliência interna do membranas (rectais prolapso retal) é mais comum, ocorrendo em até 10% das crianças com CF, e é causada pelo aumento do volume fecal, desnutrição e aumento da pressão intra-abdominal devido à tosse.
O muco espesso visto nos pulmões possui um homólogo nas secreções espessas do pâncreas, órgão responsável pelo fornecimento de sucos digestivos que ajudam a quebrar a comida. Estas secreções bloquear o movimento exócrina do enzimas digestivas para o duodeno e resultar em danos irreversíveis ao pâncreas, muitas vezes com inflamação dolorosa ( pancreatite). O ductos pancreáticos estão totalmente conectados em casos mais avançados, geralmente visto em crianças mais velhas e adolescentes. Isto faz com que a atrofia das glândulas exócrinas e fibrose progressiva.
A ausência de enzimas digestivas conduz a dificuldade em absorver nutrientes com a sua excreção subsequente nas fezes, uma doença conhecida como má absorção. Má absorção leva à desnutrição e ao crescimento e desenvolvimento pobre por causa da perda de calorias. Resultante hipoproteinemia pode ser grave o suficiente para causar edema generalizado. Indivíduos com FC também têm dificuldades de absorção das vitaminas lipossolúveis A, D , E, e K .
Para além dos problemas do pâncreas, as pessoas com experiência fibrose quística mais azia, obstrução intestinal por intussuscepção, e prisão de ventre. Os indivíduos mais velhos com fibrose cística podem desenvolver síndrome de obstrução intestinal distal quando fezes engrossado causar obstrução intestinal.
Insuficiência pancreática exócrina ocorre na maioria (85% a 90%) dos pacientes com CF. Ela está associada principalmente com "graves" mutações CFTR, onde ambos os alelos são completamente não-funcionais (por exemplo, F508 / F508). Ela ocorre em 10% a 15% dos pacientes com uma "grave" e uma mutação CFTR "suave" onde ainda há um pouco de atividade CFTR, ou onde há dois "suave" mutações CFTR. Nestes casos mais leves, há ainda a função pancreática exócrina suficiente para que a suplementação de enzima não é necessária. Geralmente, não há outras complicações GI, em fenótipos pâncreas-suficiente e, em geral, tais indivíduos geralmente tem excelente crescimento e desenvolvimento. Apesar disso, idiopática pancreatite crônica pode ocorrer em um subgrupo de indivíduos pâncreas-suficiente com FC, e está associada com dor abdominal recorrente e as complicações com risco de vida.
Secreção espessa também pode causar problemas de fígado em pacientes com CF. A bílis segregada pelo fígado para ajudar na digestão pode bloquear o ductos biliares, levando a danos no fígado. Com o tempo, isso pode levar a cicatrização e nódulos ( cirrose). O fígado não consegue livrar o sangue de toxinas e não faz importantes proteínas , tais como os responsáveis pela coagulação do sangue. A doença hepática é a terceira causa mais comum de morte associado com CF.
Endócrino
O contém o pâncreas ilhotas de Langerhans, que são responsáveis pela tomada de insulina, um hormônio que ajuda a regular o sangue glicose . Danos do pâncreas pode levar à perda da ilhota células , levando a um tipo de diabetes que é único para as pessoas com a doença. Este diabetes relacionada à fibrose cística (DMFC) compartilha características que podem ser encontrados em tipo 1 e diabéticos do tipo 2, e é uma das principais complicações não pulmonares de CF. A vitamina D está envolvida na cálcio e regulação do fosfato. Má absorção de vitamina D a partir da dieta por causa da má absorção pode conduzir à doença óssea osteoporose em que enfraqueceu os ossos são mais suscetíveis a fraturas. Além disso, as pessoas com fibrose cística freqüentemente desenvolvem discotecas de seus dedos das mãos e pés, devido aos efeitos da doença crônica e baixa de oxigênio em seus tecidos.
Infertilidade
A infertilidade afeta homens e mulheres. Pelo menos 97% dos homens com fibrose cística são inférteis, mas não estéril e pode ter filhos com técnicas de reprodução assistida. A principal causa de infertilidade em homens com fibrose cística é ausência congênita dos vasos deferentes (que normalmente liga o testículos para o dutos ejaculatórios do pênis), mas potencialmente também por outros mecanismos, tais como causando azoospermia, teratospermia e oligoasthenospermia. Muitos homens encontrados para ter a ausência congênita dos vasos deferentes durante a avaliação para a infertilidade tem uma forma leve, previamente undiagnosed da CF. Algumas mulheres têm problemas de fertilidade devido ao muco cervical ou desnutrição espessas. Em casos graves, desnutrição interrompe ovulação e causas amenorréia.
Causa
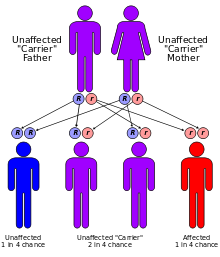
FC é causada por um mutação no gene transmembrana da fibrose cística reguladora da condutância (CFTR). A mutação mais comum, DF508, é uma deleção ( Δ eliminação significando) de três nucleótidos que resulta numa perda do aminoácido fenilalanina (F) na posição 508 na proteína. Esta mutação é responsável por dois terços (66-70%) dos casos de FC em todo o mundo e 90% dos casos no Estados Unidos ; no entanto, existem mais de 1500 outras mutações que possam produzir CF. Embora a maioria das pessoas têm duas cópias de trabalho (alelos) do gene CFTR, apenas uma é necessária para prevenir a fibrose cística. CF desenvolve quando nenhum dos alelos pode produzir uma proteína CFTR funcional. Assim, CF é considerada uma doença autossômica recessiva.
O Gene CFTR, encontrado no q31.2 locus cromossomo 7, é 230.000 pares de bases de comprimento, e cria uma proteína que é 1480 aminoácidos de comprimento. Mais especificamente, a localização é entre pares de bases 117.120.016 para 117.308.718, no braço longo do cromossomo 7, a região 3, faixa 1, sub-band 2, representado como 7q31.2. Estruturalmente, CFTR é um tipo de gene conhecido como um Gene ABC. O produto deste gene (CFTR) é um canal de iões cloreto importante na criação suor, sucos digestivos e muco. Esta proteína possui dois Hidrólise de ATP, domínios, o que permite que a proteína para utilizar a energia na forma de ATP . Ele também contém dois domínios com 6 hélices alfa cada, os quais permitem que a proteína atravessa a membrana celular. A regulamentação local de ligação na proteína permite a activação por fosforilação, principalmente pela dependente de cAMP de proteína cinase. O do terminal carboxilo da proteína é ancorada ao por um citoesqueleto Interação domínio PDZ.
Além disso, há cada vez mais evidências de que além de modificadores genéticos CFTR modula a freqüência ea gravidade da doença. Um exemplo é lectina de ligação a manana-, que está envolvida na imunidade inata, facilitando fagocitose de microrganismos. Polimorfismos em um ou ambos os alelos de lectina de ligação a manana, que resultam em baixos níveis de circulação da proteína estão associadas com um maior risco de triplo estágio final da doença do pulmão, bem como um aumento dos encargos de infecções bacterianas crónicas.
Fisiopatologia
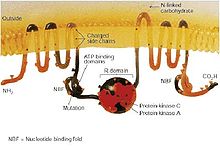
Existem várias mutações no gene CFTR, e diferentes mutações causam diferentes defeitos na proteína CFTR, por vezes, causar uma doença mais leve ou mais grave. Estes defeitos de proteínas também são alvos para drogas que podem, por vezes, restaurar a sua função. F508-CFTR, que ocorre em> 90% dos pacientes em os EUA, cria uma proteína que não faz dobrar e normalmente é degradado pela célula. Outras mutações resultam em proteínas que são muito curtos (truncada), porque a produção é encerrada prematuramente. Outras mutações produzem proteínas que não utilizam energia normalmente, não permitem cloreto, e iodeto de tiocianato de atravessar a membrana de forma adequada, ou são degradados numa taxa mais rápida do que o normal. As mutações podem também levar a menos cópias da proteína de CFTR está sendo produzida.
A proteína criada por este gene está ancorada ao membrana exterior de células no glândulas sudoríparas, pulmões, pâncreas e todas as outras glândulas exócrinas restantes no corpo. A proteína abrange a membrana e actua como um canal que liga a parte interior da célula ( citoplasma) para o fluido envolvente. Este canal é primariamente responsável por controlar a circulação de halogéneos de dentro para fora da célula; no entanto, nas condutas de suor que facilita o movimento de cloreto do suor para o citoplasma. Quando a proteína CFTR não funciona, e cloreto de tiocianato são presos no interior das células das vias aéreas e do lado de fora da pele. Em seguida hypothiocyanite, OSCN, não pode ser produzido pelo sistema de defesa imunológico. Porque cloreto é carregado negativamente , isto cria uma diferença de potencial eléctrico dentro e fora da célula causando catiões de atravessar para dentro da célula. Sódio é o catião mais comum no espaço extracelular e a combinação de cloreto de sódio e cria o sal , o qual é perdido em grandes quantidades com o suor dos indivíduos com CF. Este sal perdido constitui a base para o teste de suor.
A maioria dos danos na FC é devido ao bloqueio das passagens estreitas de órgãos afetados com secreções espessas. Estes bloqueios levar a remodelação e infecção no pulmão, os danos de acumulação de enzimas digestivas no pâncreas, obstrução do intestino grosso por fezes, etc. Existem várias teorias sobre a forma como os defeitos na proteína e a função celular causar os efeitos clínicos. Uma teoria é que a falta de halogéneo e pseudo (principalmente, cloreto, iodeto e tiocianato) que sai através da proteína CFTR leva à acumulação de muco rico em nutrientes mais viscosos nos pulmões que permite que as bactérias esconder a partir do corpo do sistema imunitário . Outra teoria é que a falta da proteína CFTR leva a um aumento paradoxal na absorção de sódio e cloreto, que, levando a um aumento da reabsorção da água, cria um muco espesso e desidratado. Contudo uma outra teoria é que o movimento de cloreto de anormal para fora da célula leva à desidratação do muco, secreções pancreáticas, secreção biliar, etc.
Infecções crônicas
Os pulmões de pacientes com fibrose cística são colonizados e infectados por bactérias a partir de uma idade precoce. Estas bactérias, que muitas vezes se espalham entre os indivíduos com CF, prosperar no muco alterado, que recolhe nas pequenas vias aéreas dos pulmões. Este muco leva à formação de micro-ambientes bacterianos conhecidos como biofilmes que são difíceis para as células do sistema imunológico e antibióticos para penetrar. Secreções viscosas e infecções respiratórias persistentes danificar repetidamente o pulmão gradualmente remodelação das vias aéreas, o que torna a infecção ainda mais difícil de erradicar.
Ao longo do tempo, ambos os tipos de bactérias e suas alterações características individuais em indivíduos com CF. Na fase inicial, as bactérias comuns, tais como o Staphylococcus aureus e Haemophilus influenzae colonizar e infectar os pulmões. Eventualmente, Pseudomonas aeruginosa (e às vezes Burkholderia cepacia) domina. Aos 18 anos de idade, 80% dos pacientes com o clássico FC porto P. aeruginosa, e 3,5% do porto B. cepacia. Uma vez dentro dos pulmões, essas bactérias se adaptar ao ambiente e desenvolver resistência aos antibióticos comumente utilizados. Pseudomonas podem desenvolver características especiais que permitem a formação de grandes colônias, conhecida como Pseudomonas "mucóides", que raramente são vistos em pessoas que não têm CF.
Uma maneira infecção se espalha é pela passagem entre diferentes indivíduos com CF. No passado, as pessoas com CF participaram muitas vezes no verão "Camps CF" e outros encontros de lazer. Hospitais pacientes com FC agrupados em áreas comuns e equipamentos de rotina (como nebulizadores) não foi esterilizada entre pacientes individuais. Isto levou a transmissão de mais estirpes de bactérias perigosas entre os grupos de pacientes. Como resultado, os indivíduos com FC são rotineiramente isolados uns dos outros no ambiente de saúde e os profissionais de saúde são encorajados a usar vestidos e luvas quando examinando pacientes com CF para limitar a propagação de estirpes de bactérias virulentas.
Pacientes com FC também podem ter suas vias aéreas cronicamente colonizados por fungos filamentosos (como Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) e / ou leveduras (tais como Cândida albicans); outros fungos filamentosos menos comumente isoladas incluem Aspergillus flavus e Aspergillus nidulans (ocorrem transitoriamente em secreções respiratórias CF), e Exophiala dermatitidis e Scedosporium prolificans (vias aéreas-colonizadores crônicas); alguns fungos filamentosos como Penicillium e emersonii Fusispora Acrophialophora são encontrados em pacientes quase exclusivamente no contexto da CF. Transporte mucociliar com defeito caracterizando CF está associado a distúrbios imunológicos locais. Além disso, o tratamento prolongado com antibióticos e da utilização de tratamentos com corticosteróides também pode facilitar o crescimento de fungos. Embora a relevância clínica da colonização das vias aéreas por fungos ainda é uma questão de debate, fungos filamentosos pode contribuir para a resposta inflamatória local, e, portanto, para a deterioração progressiva da função pulmonar, como muitas vezes acontece com aspergilose broncopulmonar alérgica (ABPA) - a doença fúngica mais comum no contexto de CF, envolvendo uma resposta imune dirigida-Th2 para Aspergillus.
Diagnóstico e monitorização
A fibrose cística pode ser diagnosticada através de vários métodos diferentes, incluindo triagem neonatal, suar teste, e testes genéticos. A partir de 2006 nos Estados Unidos, 10 por cento dos casos são diagnosticados logo após o nascimento, como parte de programas de triagem neonatal. A tela recém-nascido mede inicialmente para a concentração de sangue elevados de trypsinogen immunoreactive. Bebés com uma tela de anormal recém-nascido precisa de um teste do suor para confirmar o diagnóstico de FC. Em muitos casos, a mãe faz o diagnóstico porque a criança o gosto salgado. Tripsinogênio níveis podem ser aumentados em indivíduos que têm uma única cópia mutante do gene CFTR (transportadores) ou, em casos raros, em indivíduos com duas cópias normais do gene CFTR. Devido a estes falsos positivos, CF triagem em recém-nascidos pode ser controversa. A maioria dos estados e países não tela para CF rotina no nascimento. Portanto, a maioria dos indivíduos são diagnosticados depois que os sintomas (por exemplo, doença sinopulmonar e manifestações GI) solicitar uma avaliação para a fibrose cística. A forma mais comumente utilizada de teste é o teste do suor. Sweat-teste consiste na aplicação de um medicamento que estimula a transpiração ( pilocarpina). Para fornecer o medicamento através da pele, iontoforese é usado para, em que um eletrodo é colocado sobre a medicação aplicada e uma corrente eléctrica é passada a um eléctrodo separado sobre a pele. O suor resultante é então recolhido em papel de filtro ou num tubo capilar e analisadas para quantidades anormais de sódio e cloreto. As pessoas com FC têm aumento da quantidade de sódio e cloreto no suor. Em contraste, as pessoas com FC têm menos tiocianato e hypothiocyanite em sua saliva e muco (Banfi et al.). CF também pode ser diagnosticada através da identificação de mutações no gene de CFTR.
As pessoas com FC podem ser listados em um registro da doença que permite aos pesquisadores e médicos para acompanhar os resultados de saúde e identificar os candidatos para ensaios clínicos.
Pré-natal
Casais que estão grávidas ou a planear uma gravidez pode ter-se testado para as mutações do gene CFTR para determinar o risco que seu filho vai nascer com fibrose cística. O teste é tipicamente efectuado em primeiro lugar em um ou ambos os pais e, se o risco de FC é alta, testes sobre o feto é realizada. O Colégio Americano de Obstetras e Ginecologistas (ACOG) recomenda o teste para os casais que têm uma história pessoal ou familiar próximo da CF, e eles recomendam que o teste do portador ser oferecido a todos os casais caucasianos e disponibilizadas para os casais de outras origens étnicas.
Porque o desenvolvimento da CF no feto requer cada um dos pais para passar uma cópia mutante do gene CFTR e porque o teste CF é caro, o teste é muitas vezes realizado, inicialmente, em um dos pais. Se o teste mostra que pai é um portador da mutação do gene CFTR, o outro progenitor é testado para calcular o risco de que seus filhos terão CF. CF pode resultar a partir de mais de um milhar de diferentes mutações, a partir de 2006 e que não é possível testar para cada um. Testes analisa o sangue para as mutações mais comuns, tais como F508-ensaios disponíveis comercialmente mais olhar para 32 ou menos mutações diferentes. Se uma família tem uma mutação rara conhecida, rastreio específico para que a mutação pode ser realizada. Porque nem todas as mutações conhecidas são encontradas em testes atuais, uma tela negativo não garante que a criança não terá CF.
Durante a gravidez, os testes podem ser realizados com o placenta ( biópsia de vilo corial) ou do fluido ao redor do feto ( amniocentese). No entanto, biópsia de vilo corial tem um risco de morte fetal de 1 em 100 e amniocentese de 1 em 200; um estudo recente indicou que este pode ser mais baixo, cerca de 1 em 1,600.
Economicamente, para casais portadores de fibrose cística, quando se compara o diagnóstico genético pré-implantação (PGD) com a concepção natural (NC), seguido de teste pré-natal e do aborto de gestações afetadas, PGD fornece benefícios económicos líquidos até a idade materna de aproximadamente 40 anos, após o qual NC, testes de pré-natal e do aborto tem maior benefício econômico.
Gestão
Enquanto não há cura para a fibrose cística existem vários métodos de tratamento. A gestão da fibrose cística tem melhorado significativamente ao longo dos últimos 70 anos. Enquanto recém-nascidos com fibrose cística há 70 anos teria sido dificilmente vivem após seu primeiro ano, as crianças de hoje são propensos a viver até a idade adulta. Os recentes avanços no tratamento da fibrose cística fizeram com que um indivíduo com fibrose cística pode viver uma vida mais plena menos oneradas por sua condição. Os pilares de gestão são o tratamento pró-ativo de vias aéreas infecção, e incentivo à boa nutrição e um estilo de vida ativo. Gestão da fibrose cística continua ao longo da vida de um paciente, e visa maximizar a função do órgão e, portanto, a qualidade de vida. Na melhor das hipóteses, os tratamentos atuais atrasar o declínio da função do órgão. Devido à grande variação no tratamento da doença sintomas geralmente ocorre em centros especializados multidisciplinares, e é adaptado para o indivíduo. Alvos para a terapia são a pulmões, trato gastrointestinal (incluindo os suplementos de enzimas pancreáticas), o órgãos reprodutivos (incluindo tecnologia de reprodução assistida (ART)) e apoio psicológico.
O aspecto mais consistente do tratamento da fibrose cística é limitante e tratar a lesão pulmonar causada por muco espesso e infecção, com o objectivo de manter qualidade de vida. Intravenosa, inalado, e antibióticos orais são usados para tratar infecções crónicas e agudas. Os dispositivos mecânicos inalação e medicamentos são usados para alterar e limpar o muco engrossado. Estas terapias, embora eficazes, podem ser extremamente para o paciente consome tempo. Uma das batalhas mais importantes que os pacientes com FC enfrentar é encontrar o tempo para cumprir com os tratamentos prescritos, equilibrando uma vida normal.
Em adição, tais como terapias e transplante objectivo da terapia genética para curar alguns dos efeitos da fibrose cística. A terapia genética tem como objectivo introduzir CFTR normal das vias aéreas. Teoricamente, este processo deve ser simples como a via aérea é facilmente acessível e existe apenas um único defeito genético para corrigir. Existem dois mecanismos gene CFTR introdução envolvidos, a primeira utilização de um vector viral (adenovírus, vírus ou retro vírus adeno-associado) e em segundo lugar a utilização de lipossoma. No entanto, existem alguns problemas associados com estes métodos que envolvem a eficiência (lipossomas proteína insuficiente) e entrega (vírus provoca uma resposta imune).
Antibióticos
Muitos pacientes com FC estão em um ou mais antibióticos em todos os momentos, mesmo quando saudável, para profilaticamente suprimir a infecção. Os antibióticos são absolutamente necessárias sempre que se suspeita de pneumonia ou em que houve uma diminuição notável da função pulmonar, e são geralmente seleccionados com base nos resultados de uma análise da expectoração e da resposta do passado do paciente. Esta terapia prolongada, muitas vezes requer internação e inserção de um mais permanente IV tal como um cateter central de inserção periférica (PICC) ou Port-a-Cath. Terapia com antibióticos inalatórios, como tobramicina, colistina, e aztreonam muitas vezes é dado por meses em um tempo para melhorar a função pulmonar, impedindo o crescimento de bactérias colonizadas. Os antibióticos orais, como a ciprofloxacina ou azitromicina é dado para ajudar a prevenir a infecção ou para controlar a infecção em curso. O antibióticos aminoglicosídeos (por exemplo, tobramicina) utilizados pode causar perda, danos auditivos à sistema de equilíbrio no ouvido interno ou problemas renais com o uso a longo prazo. Para evitar que estes efeitos colaterais, a quantidade de antibióticos no sangue são rotineiramente medido e ajustado em conformidade.
Outros tratamentos para a doença pulmonar
Várias técnicas são usadas para mecânico desalojar escarro e incentivar a sua expectoração. No ambiente hospitalar, fisioterapia (CPT) é utilizado; um terapeuta respiratório percusses peito de um indivíduo com suas mãos várias vezes ao dia, para soltar secreções. Dispositivos que recriar essa terapia de percussão incluem o ThAIRapy Vest eo ventilador percussivo intrapulmonar (IPV). Métodos mais recentes, como Ventilação bifásica couraça, e associado de modo depuração disponíveis em tais dispositivos, integrar uma fase de assistência tosse, bem como uma fase de retirada de secreções vibração. Estes são portáteis e adaptado para uso doméstico.
Medicamentos em aerossol que ajudam a soltar as secreções incluem Pulmozyme® e hipertônica solução salina. A dornase é um humano recombinante deoxyribonuclease, que degrada DNA na expectoração, diminuindo assim a sua viscosidade. Denufosol é uma droga de investigação que abre um canal de cloreto alternativa, ajudando a liquefazer muco.
Como se agrava doença pulmonar, assistência respiratória mecânica pode se tornar necessário. Indivíduos com FC pode necessitar da utilização de máscaras especiais à noite que ajudam a empurrar o ar em seus pulmões. Estas máquinas, conhecidos como pressão positiva de dois níveis (BiPAP) ventiladores, ajudam a evitar baixos níveis de oxigênio no sangue durante o sono. BiPAP também pode ser utilizado durante a fisioterapia para melhorar a limpeza da secreção. Durante uma doença grave, um tubo pode ser colocado na garganta (um processo conhecido como um traqueostomia) para permitir a respiração apoiada por um ventilador.
Para as crianças que vivem com o CF, os estudos preliminares mostram pediátrica massagem terapêutica pode melhorar os pacientes e suas famílias a qualidade de vida, embora estudos mais rigorosos deve ser feito.
Transplantação
O transplante de pulmão, muitas vezes torna-se necessário que os indivíduos com fibrose cística como a função pulmonar e tolerância ao exercício diminui. Embora o transplante de pulmão único é possível em outras doenças, os indivíduos com FC deve ter dois pulmões substituído porque o pulmão remanescente pode conter bactérias que podem infectar o pulmão transplantado. Um transplante de pâncreas ou de fígado pode ser realizada ao mesmo tempo, a fim de aliviar a doença hepática e / ou diabetes. O transplante de pulmão é considerado quando a função pulmonar diminui até o ponto onde a ajuda de dispositivos mecânicos é necessário ou sobrevida do paciente está ameaçada.
Outros aspectos
Os recém-nascidos com obstrução intestinal normalmente requerem cirurgia, enquanto que adultos com síndrome de obstrução intestinal distal, normalmente, não. O tratamento da insuficiência pancreática através de substituição de enzimas digestivas em falta permite o duodeno de absorver adequadamente nutrientes e vitaminas que poderiam ser perdidos no feces.So agora, nenhuma investigação em grande escala envolvendo a incidência de aterosclerose e doença cardíaca coronária em adultos com fibrose cística tem sido conduzido. Isto é provavelmente devido ao facto de a grande maioria das pessoas com fibrose cística não viver o tempo suficiente para desenvolver aterosclerose clinicamente significativa ou doença cardíaca coronária.
A diabetes é a complicação não-pulmonar mais comum de CF. Ele combina características de tipo 1 e diabetes tipo 2, e é reconhecido como uma entidade distinta, diabetes relacionada à fibrose cística (DMFC). Enquanto via oral fármacos anti-diabéticos são por vezes usados, o único tratamento recomendado é a utilização de insulina ou um injecções bomba de insulina, e, ao contrário do tipo 1 e 2 diabetes, restrições alimentares não são recomendadas.
Desenvolvimento de osteoporose pode ser prevenida por aumento da ingestão de vitamina D e cálcio , e podem ser tratados por bifosfonatos, embora efeitos adversos podem ser um problema. Crescimento deficiente pode ser evitada por meio da inserção de um tubo de alimentação para o aumento calorias através suplementares feeds ou por administração de injetada hormona de crescimento.
Seio infecções são tratadas por meio de cursos prolongada de antibióticos. O desenvolvimento de pólipos nasais ou outras alterações crónicas no interior das passagens nasais, podem limitar severamente o fluxo de ar através do nariz, e ao longo do tempo reduzir sentido de cheiro do paciente. Cirurgia da cavidade é muitas vezes usado para aliviar a obstrução nasal e limitar novas infecções. Esteróides nasais como fluticasona são usados para diminuir a inflamação nasal.
Infertilidade feminina pode ser superada por tecnologia de reprodução assistida, particularmente técnicas de transferência de embriões. Infertilidade masculina causada pela ausência do vasos deferentes podem ser superados com extracção testicular de espermatozóides (TEST), coleta de espermatozóides diretamente dos testículos. Se a amostra coletada contém muito poucos espermatozóides para ter um provável espontânea fertilização, injeção intracitoplasmática de espermatozóides pode ser realizada. Reprodução de terceiros também é uma possibilidade para as mulheres com CF.
Prognóstico
O prognóstico para a fibrose cística melhorou devido a um diagnóstico precoce através da triagem, tratamento e melhor acesso aos cuidados de saúde. Em 1959, a idade mediana de sobrevida de crianças com fibrose cística nos Estados Unidos foi de seis meses. Em 2008, a sobrevida média de 37,4 anos. No Canadá, a média de sobrevida aumentou de 24 anos em 1982-47,7 em 2007.
Daqueles com fibrose cística que são mais de 18 anos a partir de 2009; 92% se formou na escola secundária, 67% tinham pelo menos alguma educação universitária, 15% foram desativados e 9% estavam desempregados, 56% eram solteiros e 39% eram casados ou viviam com um parceiro. Em Rússia a idade média dos pacientes é em geral 25, que é causada pela ausência ou alto custo da medicação e o facto de que o transplante de pulmão não é realizada.
Qualidade de vida
As doenças crônicas podem ser muito difícil de gerir. A fibrose cística (FC) é uma doença crônica que afeta os "aparelhos digestivos e respiratórios resultando em desnutrição generalizada e infecções respiratórias crónicas". As secreções espessas obstruir as vias aéreas nos pulmões, o que muitas vezes causam inflamações e infecções pulmonares graves. Portanto, o muco torna difícil para respirar. Se ele for comprometido, isso afeta a qualidade de vida de alguém com CF, e sua capacidade para completar tarefas como tarefas cotidianas. É importante que os pacientes com FC para compreender a relação prejudicial que as doenças crónicas colocar sobre a qualidade de vida. De acordo com Schmitz e Goldbeck (2006), o fato de que a fibrose cística aumenta significativamente o estresse emocional, tanto do indivíduo e da família ", ea rotina de tratamento diário necessário demorado pode ter efeitos ainda mais negativos sobre a qualidade de vida (QV)". No entanto, Havermans e colegas (2006) têm mostrado que jovens pacientes ambulatoriais com CF que tenham participado no CFQ-R (Cystic Fibrosis Questionnaire-revista) "classificado alguns domínios de qualidade de vida mais elevada do que os seus pais". Conseqüentemente, pacientes ambulatoriais com FC têm uma visão mais positiva para si próprios. Além disso, existem muitas maneiras de melhorar a qualidade de vida em pacientes com FC. Exercício é promovido para aumentar a função pulmonar. O facto de integrar um regime de exercícios na rotina diária do paciente CF pode melhorar significativamente a qualidade de vida. Não há cura definitiva para a fibrose cística. No entanto, há diversos medicamentos utilizados, tais como, mucolíticos, broncodilatadores, esteróides e antibióticos que têm o efeito de afrouxamento do muco das vias aéreas, em expansão, diminuindo a inflamação e combate a infecções pulmonares.
Epidemiologia
| Mutação | Freqüência mundial |
|---|---|
| F508 | 66% -70% |
| G542X | 2,4% |
| G551D | 1,6% |
| N1303K | 1,3% |
| W1282X | 1,2% |
| Todos os outros | 27,5% |
A fibrose cística é a autossômica limitando-vida doença recessiva mais comum entre as pessoas de herança caucasiana. Nos Estados Unidos, aproximadamente 30.000 indivíduos têm CF; a maioria são diagnosticados por seis meses de idade. No Canadá , existem cerca de 3.500 pessoas com CF. Cerca de 1 em 25 pessoas de ascendência europeia, e um em 30 dos americanos caucasianos, é portadora de uma mutação da fibrose cística. Embora CF é menos comum nestes grupos, aproximadamente 1 em 46 hispânicos, um em 65 africanos e 1 em 90 asiáticos têm pelo menos um gene CFTR anormal. A Irlanda tem maior incidência do mundo de fibrose cística, em 1: 1353.
Embora tecnicamente um doença rara, a fibrose cística é classificado como uma das doenças genéticas mais difundidos vida encurtamento. É mais comum entre as nações do mundo ocidental. Uma exceção é a Finlândia , onde apenas um em cada 80 pessoas portadoras de uma mutação FC. Nos Estados Unidos, 1 em cada 4.000 crianças nascem com CF. Em 1997, cerca de 1 em 3.300 caucasiano crianças nos Estados Unidos nasceu com fibrose cística. Em contraste, apenas 1 em cada 15 mil crianças americanas africanas sofria de fibrose cística, e em americanos de origem asiática a taxa foi ainda menor em 1 em 32.000.
A fibrose cística é diagnosticada em homens e mulheres igualmente. Por razões que permanecem obscuras, os dados mostraram que os machos tendem a ter uma maior expectativa de vida do que as fêmeas, porém estudos recentes sugerem que esta diferença de gênero pode não existir mais, talvez, devido a melhorias em instalações de cuidados de saúde, enquanto um estudo recente da Irlanda identificou um link entre o hormônio feminino estrogênio e piores resultados em CF.
A distribuição dos alelos FC varia entre as populações. A freqüência de portadores DF508 foi estimada em 1: 200 no norte da Suécia, 1: 143 em lituanos, e 01:38 na Dinamarca. Nenhum transportadoras DF508 foram encontrados entre 171 finlandeses e 151 Sami pessoas. DF508 ocorre na Finlândia, mas é um alelo minoria lá. A fibrose cística é conhecido para ocorrer em apenas 20 famílias (pedigrees) na Finlândia.
Hipóteses sobre a prevalência
O Mutação DF508 é estimado em até 52.000 anos de idade. Inúmeras hipóteses têm sido avançadas a respeito de porque uma mutação tão letais tem persistido e se espalhou na população humana. Outras doenças autossômicas recessivas comum, tais como anemia falciforme foram encontrados para proteger portadores de outras doenças, um conceito conhecido como vantagem heterozigoto. Resistência à seguinte foram propostas como possíveis fontes de vantagem heterozigoto:
- Cólera: Com a descoberta de que a toxina da cólera exige host normal CFTR proteínas para funcionar corretamente, se a hipótese de que os portadores de genes CFTR mutante beneficiou de resistência à cólera e outras causas de diarréia. Estudos posteriores não confirmaram essa hipótese.
- Febre tifóide: proteínas CFTR normal são também essenciais para a entrada de Salmonella Typhi nas células, o que sugere que os portadores de genes CFTR mutante pode ser resistente a febre tifóide . Sem in vivo estudo confirmou ainda esta. Em ambos os casos, o baixo nível de fibrose cística fora da Europa, em locais onde tanto a cólera ea febre tifóide é endêmica, não é imediatamente explicável.
- Diarréia: Foi também a hipótese de que a prevalência da CF na Europa pode estar relacionada com o desenvolvimento do gado de domesticação. Nesta hipótese, portadores de um único cromossoma CFTR mutante tinha alguma proteção contra a diarreia causada por intolerância à lactose, antes do aparecimento das mutações que criou a tolerância à lactose.
- Tuberculose: Outra explicação possível é que os portadores do gene poderia ter alguma resistência à TB.
História

Supõe-se que a FC apareceu cerca de 3.000 aC por causa da migração dos povos, as mutações genéticas, e novas condições em nutrição. Apesar de todo o espectro clínico da CF não foi reconhecido até a década de 1930, alguns aspectos da CF foram identificados muito mais cedo. Na verdade, a literatura da Alemanha e da Suíça no século 18 advertiu Wehe dem Kind, das beim Kuss auf die Stirn salzig schmekt, er ist und muss verhext sterbe careca ou "Ai de a criança que gosto salgado de um beijo na testa, para ele é amaldiçoado e em breve deve morrer ", reconhecendo a associação entre a perda de sal na CF e na doença.
No século 19, Carl von Rokitansky descreveu um caso de morte fetal com peritonite meconial, uma complicação do íleo meconial associada com a fibrose cística. O íleo meconial foi descrito primeiramente em 1905 por Karl Landsteiner. Em 1936, Guido Fanconi publicou um artigo descrevendo a conexão entre a doença celíaca, fibrose cística do pâncreas, e bronquiectasias.
Em 1938 Dorothy Hansine Andersen publicou um artigo, "Fibrose cística do pâncreas e sua relação com a doença celíaca: um estudo clínico e patológico", no American Journal of Doenças da Infância . Ela foi o primeiro a descrever a característica da fibrose cística do pâncreas e correlacioná-la com o pulmão e doença intestinal proeminente na CF. Ela também a primeira hipótese de que a CF foi uma doença recessiva e usado pela primeira vez reposição de enzimas pancreáticas para tratar crianças afetadas. Em 1952, Paul di Sant 'Agnese descobriram anomalias em suor eletrólitos; um teste do suor foi desenvolvido e melhorado ao longo da próxima década.
A primeira ligação entre CF e outro marcador (Paroxonase) foi encontrada em 1985, indicando que apenas um locus existe para CF Hans Hans. Em 1988, a primeira mutação de CF, F508 foi descoberto por Francis Collins, Lap-Chee Tsui e John R. Riordan no sétimo cromossomo. Pesquisas posteriores encontrou mais de 1.000 mutações diferentes que causam CF.
Porque as mutações no gene CFTR são tipicamente pequenos, técnicas de genética clássica tinha sido incapaz de localizar com precisão o gene mutado. Utilizar marcadores de proteína, os estudos de ligação genética foram capazes de mapear a mutação no cromossoma 7. Cromossoma-curta e técnicas -jumping foram então usadas para identificar e sequenciar o gene. Em 1989 Lap-Chee Tsui liderou uma equipe de pesquisadores do Hospital for Sick Children, em Toronto , que descobriu o gene responsável pela CF. A fibrose cística representa a primeira doença genética estritamente elucidado pelo processo de genética inversa.
Pesquisa
A terapia génica
A terapia genética tem sido explorada como uma cura potencial para a fibrose cística. Idealmente, a terapia genética coloca uma cópia normal do gene CFTR para as células afectadas. A transferência do gene CFTR normal para as células do epitélio afetado resultaria na produção de CFTR funcional em todas as células-alvo, sem reacções adversas ou uma resposta inflamatória. Estudos têm mostrado que, para prevenir as manifestações pulmonares da fibrose cística, apenas 5-10% da quantidade normal de CFTR a expressão do gene é necessária. Várias abordagens têm sido testados para a transferência de genes, tais como lipossomas e vectores virais em modelos animais e em ensaios clínicos. No entanto, ambos os métodos foram encontrados para as opções de tratamento ser relativamente ineficientes. A razão principal é que muito poucas células tomam-se o vector e expressar o gene, de modo que o tratamento tem pouco efeito. Além disso, têm sido observados problemas no ADNc de recombinação, de tal modo que o gene introduzido por tratamento é tornada inutilizável. Com a ajuda da Fibrose Cística Trust, que tem uma liga de terapeutas de genes altamente profissionais, tanto somática e vetor viral adeno-associado fizeram avanços. O Adenoviridae, ou mais comumente conhecido como o vírus do resfriado, é geneticamente alterado, permitindo que o gene CFTR para entrar nas células pulmonares.
Pequenas moléculas
Um número de pequenas moléculas que visam compensar várias mutações do gene CFTR estão em desenvolvimento. Uma abordagem é desenvolver drogas que recebem o ribossoma para superar o codão de paragem e sintetizar uma proteína de CFTR de comprimento completo. Cerca de 10% de CF resultar de um codão de paragem prematuro no ADN, levando a terminação precoce da síntese de proteínas e proteínas truncadas. Estas drogas alvo , tais como mutações nonsense G542X, que consiste em o aminoácido glicina na posição 542 sendo substituída por um codão de paragem. Antibióticos aminoglicosídicos interferir com a síntese de ADN e de correcção de erros. Em alguns casos, eles podem causar a célula para superar o codão de paragem, inserir um aminoácido aleatório, e expressar uma proteína de comprimento completo. O aminoglicósido gentamicina tem sido utilizada para tratar as células do pulmão de pacientes com FC no laboratório para induzir as células a crescer proteínas de comprimento total. Outra droga segmentação mutações nonsense é ataluren, que está passando por fase III de ensaios clínicos a partir de outubro de 2011.
Ivacaftor (Kalydeco), aprovado para uso pelo FDA nos Estados Unidos em janeiro de 2012, tem como alvo a mutação G551D (glicina na posição 551 é substituído porácido aspártico).Lumacaftor visaF508del (fenilalanina na posição 508 está ausente).